
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe saturation of the gas phase acidity of nHF/AlF3 and nHF/GeF4 (n = 1–6) superacids caused by increasing the number of surrounding HF molecules†
Marcin
Czapla
a,
Iwona
Anusiewicz
ab and
Piotr
Skurski
*ab
aLaboratory of Quantum Chemistry, Faculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland. E-mail: piotr.skurski@ug.edu.pl
bDepartment of Technical Physics and Applied Mathematics, Gdańsk University of Technology, Narutowicza 11/12, 80-233 Gdańsk, Poland
First published on 15th March 2016
Abstract
The acidic strength of selected Brønsted/Lewis superacids is evaluated on the basis of theoretical calculations carried out at the QCISD/6-311++G(d,p) level. The energies and Gibbs free energies of deprotonation processes for nHF/AlF3 and nHF/GeF4 (n = 1–6) are found to depend on the number (n) of hydrogen fluoride molecules (playing a Brønsted acid role) surrounding the AlF3 and GeF4 Lewis acids. The successive attachment of HF molecules to either AlF3 or GeF4 gradually increases the acidity strength of the resulting superacid, which leads to the saturation achieved for 5–6 HF molecules interacting with either one of these Lewis acids. The importance of the microsolvation of the corresponding anionic species as well as the necessity of considering larger (more structurally complex) building blocks of superacids while predicting their acidity is indicated and discussed.
1. Introduction
Superacids are commonly considered as compounds exhibiting acidity stronger than 100% sulfuric acid, which means that their Hammett acidity function (H0) is smaller than −12.1,2 Even though this term was used for the very first time in 1927,3 the superacid chemistry was developed mainly in the 1960s and 1970s by Olah and Hogeveen, who investigated non-aqueous HSO3F/SbF5 and HF/SbF5 systems,4–9 and by Gillespie.1,2 Since then, superacids remain the subject of continuing theoretical10–13 and experimental14–19 investigations concerning their structure, stability and acidity. Our group contributed to these studies by addressing the issue of the HAlCl4 instability,20 predicting the acidic strength of the aluminum-based HF/AlF3 (HAlF4), HF/Al2F6 (HAl2F7), HF/Al3F9 (HAl3F10), and HF/Al4F12 (HAl4F13) systems,21 investigating the dissociative excess electron attachment to the HAlF4 superacid22 (whose properties were earlier determined by the Radom group23,24), examining the strength of the Brønsted/Lewis superacids containing In, Sn, and Sb (i.e., HInnF3n+1, HSnnF4n+1, and HSbnF5n+1 (n = 1–3)),25 and, most recently, by demonstrating that the protonation of superhalogen anions26,27 might be considered as the route to superacids' formation in selected cases only,28 despite the fact that various superhalogens containing heavy metals as central atoms (e.g., InF4, SbF6, Sb2F11, SnF5, Sn2F9) were utilized in the past to create atypical salts and complexes29–35 even with noble gases (Kr and Xe).36–39The Lewis–Brønsted superacids consist of strong Lewis acid molecules (such as AlF3) interacting with strong Brønsted acid molecules (e.g., HF) and thus their deprotonation process might be described by the following reaction scheme (that assumes the excess of a representative Brønsted acid):
| nHF/AlF3 → ((n − 1)HF/AlF4)− + H+. |
Clearly, the microsolvation of an anionic species (whose role is played by the AlF4− in the above scheme) is expected to be responsible for the change in energy with respect to the neutral microsolvated species. The Gibbs free energies of the superacid deprotonation reactions (ΔGacid) are commonly utilized while describing the acidity of superacids. Although estimated only for the gas phase, the ΔGacid values were found useful in designing novel systems exhibiting significant acidity. It is worth noting that the strongest superacids proposed thus far were found to possess their Gibbs free energies of deprotonation in the 249–270 kcal mol−1 range.11 Most recently, Srivastava and Misra also reported small ΔGacid values (indicating strong acidity) for HBeCl3 (272 kcal mol−1), HPF6 (281 kcal mol−1), and HLiCl2 (284 kcal mol−1),13 whereas our group demonstrated that even smaller Gibbs free deprotonation energies are estimated for HGaCl4 (265 kcal mol−1),28 HSn3F13 (244 kcal mol−1),25 HAl4F13 (249 kcal mol−1),21 and HSb3F16 (230 kcal mol−1).25 In fact, the last presented ΔGacid value of 230 kcal mol−1 predicted for the HF/Sb3F15 represents25 the smallest gas phase Gibbs free energy of deprotonation reported in the literature thus far (including the corresponding values characterizing F(SO3)4H and HSbF6 superacids).11 Albeit the existence of the HSb3F16 superacid has not yet been confirmed experimentally, its deprotonated (i.e., anionic) form Sb3F16− is a well-known system which was extensively utilized to create atypical salts and complexes.40–42
As indicated above, the theoretical search for novel superacids that have been carried out during last few years led to proposing various promising molecular systems whose usefulness as strong acids is yet to be verified experimentally. In our opinion, however, one important issue was being neglected while performing those investigations employing quantum chemistry methods. Namely, it was preconceived that the number of molecules playing the Brønsted acid role (e.g., HF) is approximately equal to the number of molecules that play the Lewis acid role (e.g., SbF5, AlF3) in the mixture that represents a given Lewis–Brønsted superacid. In other words, it was assumed that these both components are combined using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. As a consequence, the simplest ‘building block’ that was supposed to exhibit the superacid properties was thought of as composed of a single Lewis acid molecule interacting with one Brønsted acid system. Thus, in this contribution we are going to address the issue of the Brønsted/Lewis system acidity in a different way, namely, we intend to verify whether the acidic strength of such species depends on the number of Brønsted acid molecules surrounding a single Lewis acid moiety. Our decision to undertake such a study was motivated by the following observations: (i) the recently reported crystal structures of the HF/AsF5 (HAsF6)19 and HF/SbF5 (HSbF6)15 superacids clearly show the presence of more than one HF molecule in the vicinity of the Lewis acid unit (either AsF5 or SbF5); (ii) earlier experimental studies revealed the existence of various ions (i.e., H2F+, H3F2+, SbF6−) in the liquid HF/SbF5 (HSbF6);43–45 and (iii) some superacid preparation procedures describe the use of the excess of anhydrous hydrogen fluoride.19 Therefore, one might speculate that each Lewis acid molecule may interact with more than one HF moiety in the final superacid mixture. In addition, it seems likely that the mutual interactions among the HF moieties surrounding each Lewis acid molecule contribute to the system's ability to donate a proton. Hence, in order to shed more needed light on this problem, we decided to investigate the gas phase Gibbs free deprotonation energy dependence on the number of hydrogen fluoride molecules surrounding two arbitrarily chosen Lewis acids (i.e., AlF3 and GeF4).
:1 ratio. As a consequence, the simplest ‘building block’ that was supposed to exhibit the superacid properties was thought of as composed of a single Lewis acid molecule interacting with one Brønsted acid system. Thus, in this contribution we are going to address the issue of the Brønsted/Lewis system acidity in a different way, namely, we intend to verify whether the acidic strength of such species depends on the number of Brønsted acid molecules surrounding a single Lewis acid moiety. Our decision to undertake such a study was motivated by the following observations: (i) the recently reported crystal structures of the HF/AsF5 (HAsF6)19 and HF/SbF5 (HSbF6)15 superacids clearly show the presence of more than one HF molecule in the vicinity of the Lewis acid unit (either AsF5 or SbF5); (ii) earlier experimental studies revealed the existence of various ions (i.e., H2F+, H3F2+, SbF6−) in the liquid HF/SbF5 (HSbF6);43–45 and (iii) some superacid preparation procedures describe the use of the excess of anhydrous hydrogen fluoride.19 Therefore, one might speculate that each Lewis acid molecule may interact with more than one HF moiety in the final superacid mixture. In addition, it seems likely that the mutual interactions among the HF moieties surrounding each Lewis acid molecule contribute to the system's ability to donate a proton. Hence, in order to shed more needed light on this problem, we decided to investigate the gas phase Gibbs free deprotonation energy dependence on the number of hydrogen fluoride molecules surrounding two arbitrarily chosen Lewis acids (i.e., AlF3 and GeF4).
2. Methods
The nHF/AlF3 and nHF/GeF4 (n = 1–6) closed-shell neutral systems (i.e., AlF3 and GeF4 Lewis acids surrounded by n hydrogen fluoride molecules) and their corresponding anions (i.e., negatively charged closed-shell species formed by deprotonation) were investigated using theoretical quantum chemistry methods. In particular, the equilibrium geometries and harmonic vibrational frequencies were calculated using Density Functional Theory (DFT) method with the B3LYP46,47 functional and the 6-311++G(d,p)48,49 basis sets. The final electronic energies of all such determined structures were obtained by applying the quadratic configuration interaction method with single and double substitutions (QCISD)50–52 together with the same 6-311++G(d,p) basis set. The predicted error in estimating deprotonation energy values due to the 6-311++G(d,p) basis set choice was estimated as not exceeding 2 kcal mol−1 (which was evaluated by comparing the DE values obtained with the 6-311++G(d,p) and aug-cc-pVTZ basis sets for one representative superacid).The random search was performed while exploring the configuration space of each nHF/AlF3 and nHF/GeF4 (n = 1–6) closed-shell neutral as well as ((n − 1)HF/AlF4)− and ((n − 1)HF/GeF5)− (n = 1–6) closed-shell anionic system. Namely, in the case of neutral systems, various possibilities of attaching the HF molecules to either AlF3 or GeF4 were examined by treating them as the starting structures during the independent geometry optimization procedures. In addition, various combinations of mutual interactions among the HF systems were taken into account. In the case of negatively charged systems, our search was mostly based on assuming the presence of a central unit (either AlF4 or GeF5) in the structure (as the AlF4− or GeF5− correspond to very strongly bound anions).
The electronic and Gibbs free energies of the deprotonation reactions (DE and ΔGacid, respectively) were evaluated using the QCISD electronic energies and the zero-point energy corrections, thermal corrections (at T = 298.15 K) and entropy contributions estimated with the B3LYP method and 6-311++G(d,p) basis set (in each case the Gibbs free energy of the proton was also accounted for). The resulting ΔGacid values correspond to the Gibbs free energies characterizing the following processes for (n = 1 − 6):
| nHF/AlF3 → ((n − 1)HF/AlF4)− + H+ |
| nHF/GeF4 → ((n − 1)HF/GeF5)− + H+. |
Since the proper evaluation of the thermodynamic properties might be questionable in the case of weakly bound systems, we mainly focus on the deprotonation energies (DE) characterizing the species investigated, whereas the presented Gibbs free deprotonation energies (ΔGacid) should be considered as less reliable and possibly plagued by errors.
The partial atomic charges (qESP) were fitted to the electrostatic potential according to the Merz–Singh–Kollman scheme.53
All calculations were performed using the GAUSSIAN 09 (Rev. A.02) package.54
3. Results
3.1. The AlF3 Lewis acid surrounded by various number of HF molecules
The lowest energy structures of nHF/AlF3 (n = 1–6) systems are depicted in Fig. 1 whereas the corresponding higher energy isomers are shown in Fig. 2. The simplest case of the AlF3 Lewis acid interacting with one HF molecule, HF/AlF3 (HAlF4), has already been characterized as a superacid composed of the hydrogen fluoride donating its fluorine's lone pair to the empty Al's 3p atomic orbital of AlF3 quasi-planar fragment and additionally stabilized by the FH⋯F3Al hydrogen bond.21,22,28 The DE of 279–280 kcal mol−1 and ΔGacid of 267–269 kcal mol−1 (depending on the theory level employed)21,22,28 were predicted for this superacid, however, in this contribution we assume the DE = 279 kcal mol−1 and ΔGacid = 267 kcal mol−1 values for consistency with the results presented for the remaining nHF/AlF3 (n = 2–6) systems. The 2HF/AlF3 species might be viewed as formed by the attachment of the second HF molecule to the HF/AlF3 system, hence it resembles the deformed neutral AlF3 molecule with two HF moieties attached, see structure 2HF/AlF3 (1) in Fig. 1. One HF molecule forms a dative HF → AlF3 bond (1.919 Å) with the AlF3 whereas the other HF fragment is involved in the formation of two H-bonds. The DE of 278 kcal mol−1 and ΔGacid of 264 kcal mol−1 were predicted for this system which indicates the decrease of DE by 1 kcal mol−1 and the decrease of ΔGacid by 3 kcal mol−1 with respect to the HF/AlF3, see Table 1. In addition, we found two other isomeric structures of 2HF/AlF3 (see 2HF/AlF3 (2) and 2HF/AlF3 (3) in Fig. 2) having their energy larger by 6–9 kcal mol−1 than the lowest energy structure 2HF/AlF3 (1). Interestingly, the structures of these higher energy isomers are qualitatively different, as the isomer 2HF/AlF3 (2) contains two HF molecules localized on the opposite sides of the quasi-planar AlF3 fragment which allows for the formation of two dative bonds (HF → Al(3p) ← FH) and two H-bonds between HF and AlF3 fragments, whereas the structure of 2HF/AlF3 (3) contains one HF system forming a dative HF → Al(3p) bond with AlF3 and the second one linked (via the H-bond) to the fluorine atom of the central AlF3 unit, see Fig. 2.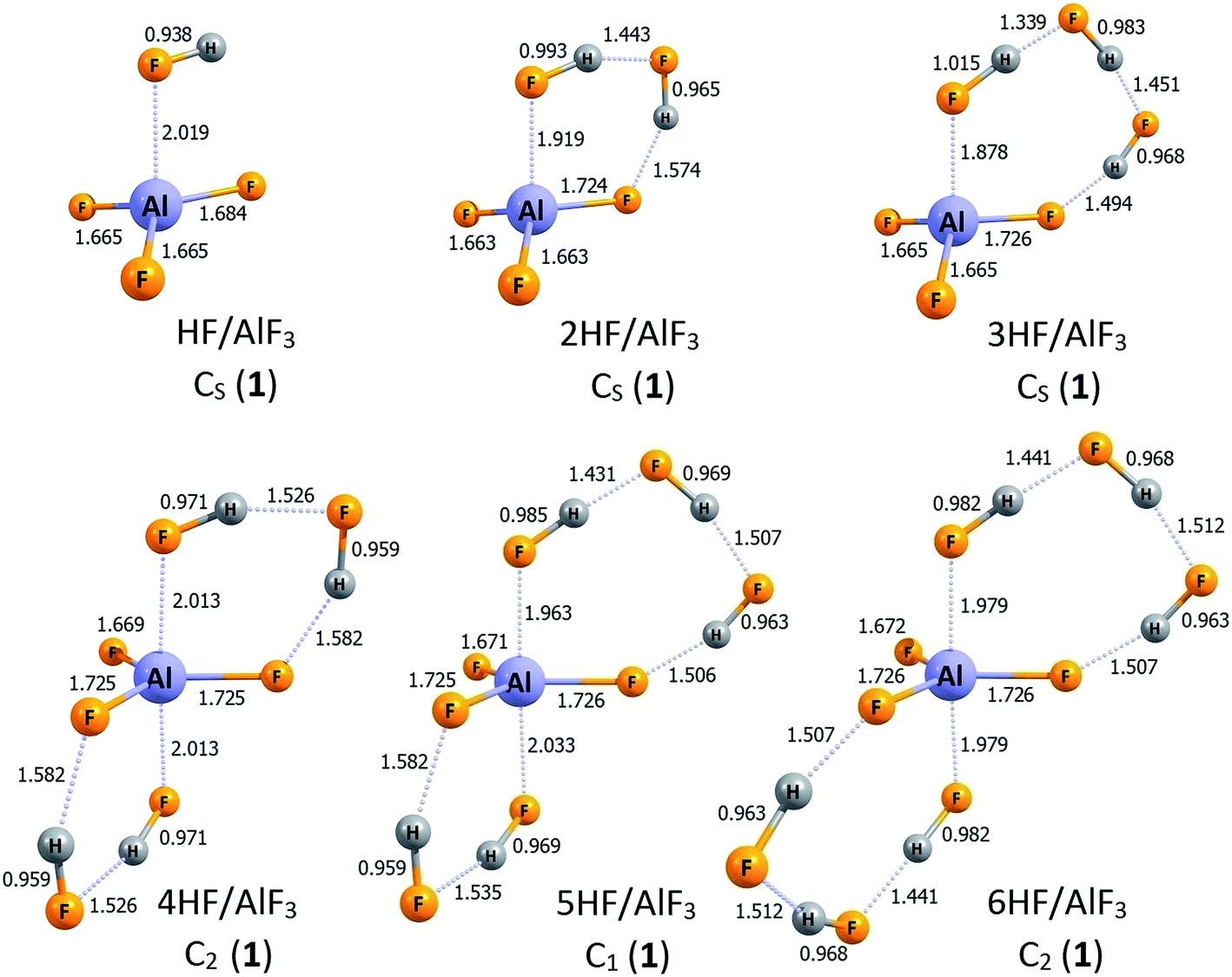 | ||
| Fig. 1 The structures of the lowest energy isomers of the nHF/AlF3 superacids (n = 1–6). Selected bond lengths are given in Å. Dative bonds and hydrogen bonds are represented by the dotted lines. | ||
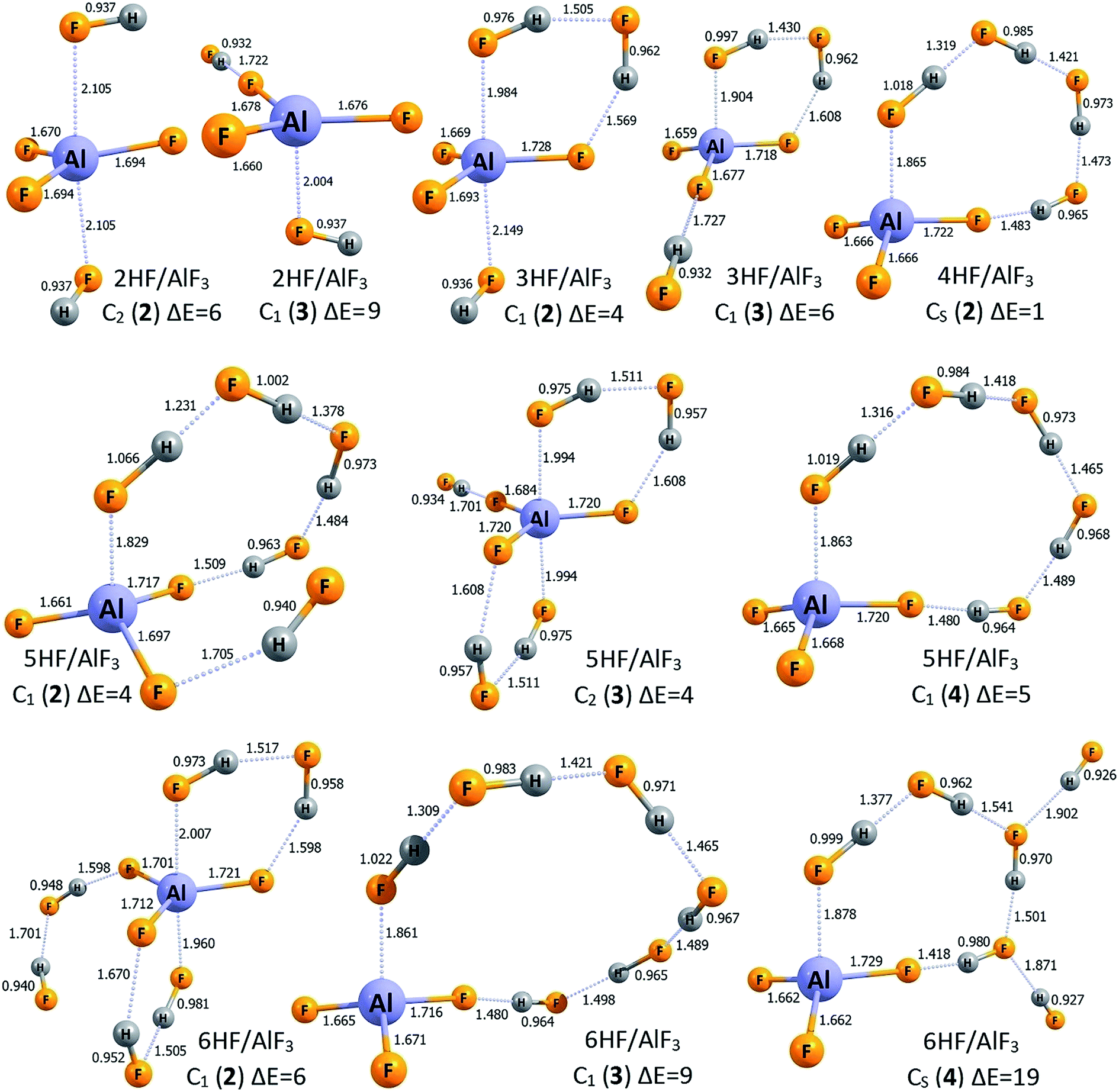 | ||
| Fig. 2 The structures of the higher energy isomers of the nHF/AlF3 superacids (n = 1–6). Selected bond lengths are given in Å. Relative energies (ΔE) with respect to the corresponding lowest energy isomers are given in kcal mol−1. Dative bonds and hydrogen bonds are represented by the dotted lines. | ||
| Species | Symmetry | DE | TΔS | ΔGacid |
|---|---|---|---|---|
| HF/AlF3 | C s | 279.2 | 6.3 | 267.4 |
| 2HF/AlF3 | C s | 277.8 | 8.6 | 264.3 |
| 3HF/AlF3 | C s | 274.6 | 9.6 | 260.8 |
| 4HF/AlF3 | C 2 | 270.5 | 12.8 | 253.1 |
| 5HF/AlF3 | C 1 | 268.9 | 13.4 | 251.3 |
| 6HF/AlF3 | C 2 | 270.4 | 15.3 | 251.1 |
The lowest energy isomer 3HF/AlF3 (1) depicted in Fig. 1 resembles the most stable structure of 2HF/AlF3 (1) with one more HF molecule involved in the resulting three member HF-based bridge whose F-end forms a dative bond with Al's empty 3p atomic orbital while the H-terminus is linked (via H-bond) to the F atom of the AlF3 fragment. It seems important to stress that the H–F bond lengths in three HF molecules span the 0.968–1.015 Å range and the partial atomic charges in each of these HF units sum up to zero which clearly indicates that the whole system is correctly described by the 3HF/AlF3 formula, see Fig. 1. The acidity of 3HF/AlF3 (1) is slightly stronger than that of 2HF/AlF3 (1) as its DE of 275 kcal mol−1 is smaller by 3 kcal mol−1 than the corresponding DE characterizing the 2HF/AlF3 (1), see Table 1 (analogous decrease of ΔGacid by 3 kcal mol−1 is also observed). We also found two other geometrically stable structures of 3HF/AlF3 (depicted in Fig. 2 as 3HF/AlF3 (2) and 3HF/AlF3 (3)) having their electronic energies within 6 kcal mol−1 with respect to the global minimum. The structure 3HF/AlF3 (2) resembles the 2HF/AlF3 (2) with the additional (third) HF molecule enabling the H-bond connection to the AlF3 moiety, whereas the structure of 3HF/AlF3 (3) clearly corresponds to the 2HF/AlF3 (1) with the additional HF molecule H-bonded to the fluorine atom of the AlF3, see Fig. 1 and 2. Again, it appears that all three HF fragments in both 3HF/AlF3 (2) and 3HF/AlF3 (3) are typical hydrogen fluoride systems with their vanishing net partial atomic charges and 0.932–0.997 Å bond lengths.
The lowest energy structure of 4HF/AlF3 (depicted in Fig. 1 as 4HF/AlF3 (1)) mimics a trigonal bipyramid AlF5 having two elongated (2.013 Å) F → Al dative bonds and three shorter (1.669–1.725 Å) covalent Al–F bonds (in the planar AlF3 fragment) whose fluorine ligands are connected via the H-bond networks. However, taking into account the bond lengths and partial atomic charges (summing up to zero for each of four HF fragments), one should consider this system as the AlF3 and four HF molecules assembled together (rather than, for instance, composed of the AlF52− dianion and two H2F+ cations). As far as the acidity of the 4HF/AlF3 (1) is concerned, we predicted the DE of 271 kcal mol−1 (ΔGacid = 253 kcal mol−1), which indicates that the acidic strength of this system should be even larger than the acidity of AlF3 surrounded by a smaller number (from 1 to 3) HF molecules, see Table 1. We also found one higher energy isomer of this species (depicted in Fig. 2 as 4HF/AlF3 (2)) whose relative energy is larger by only 1 kcal mol−1. Such a quasi-degeneracy clearly indicates that both of these structures (i.e., 1 and 2 of 4HF/AlF3) should be present in the bulk. The isomer 4HF/AlF3 (2), however, possesses a different structure than 4HF/AlF3 (1), as it corresponds to the quasi-planar AlF3 unit whose Al atom and F atom are connected with the (HF)4 H-bonded linkage (analogously to the global minimum of 3HF/AlF3 where similar (HF)3 internally H-bonded linkage is present, see 3HF/AlF3 (1) in Fig. 1). In addition, the conclusion about the presence of the AlF3 and four HF units formulated for the structure 4HF/AlF3 (1) (i.e., indicating the absence of H2F+ cations) remains valid also for the competitive isomer 4HF/AlF3 (2), hence justifying its 4HF/AlF3 formula.
While investigating the AlF3 Lewis acid surrounded by five HF molecules, 5HF/AlF3, we found the lowest energy structure 5HF/AlF3 (1) (depicted in Fig. 1) and three higher energy isomers (shown in Fig. 2 as isomers 2, 3, and 4). The global minimum 5HF/AlF3 (1) resembles the lowest energy isomer 4HF/AlF3 (1) as it also contains a trigonal bipyramid AlF5 with two elongated (1.963–2.033 Å) F → Al dative bonds and three shorter (1.671–1.726 Å) covalent Al–F bonds (in the planar AlF3 fragment) whose F ligands are linked via the H-bonded units. However, as it was observed and discussed for 4HF/AlF3 (1) (see the preceding paragraph), the more proper way of viewing this system is that consistent with the 5HF/AlF3 formula, as neither AlF52− dianion nor H2F+ cations are present (as it might have been expected when distinguishing the AlF5 core as the independent fragment of the structure). Such a conclusion is supported by the typical (0.959–0.985 Å) lengths of H–F bonds in all five HF moieties and their vanishing net atomic partial charges (i.e., atomic partial charges in each HF fragment sum up to zero and this is also the case for the remaining AlF3 fragment), see Fig. 1. The DE of 269 kcal mol−1 was predicted for 5HF/AlF3 (1) which means the 2 kcal mol−1 decrease in comparison to 4HF/AlF3 (1), see Table 1. Certainly, it should result in a slightly stronger acidity of the former compound, however the reported DE drop seems rather small. As far as the higher energy isomers of 5HF/AlF3 are concerned, the three remaining isomers shown in Fig. 2 possess their relative energies within 4–5 kcal mol−1 and thus they might be considered as competitive. The structure of 5HF/AlF3 (2) resembles that of 4HF/AlF3 (2) with the additional HF molecule attached to the different fluorine atom of AlF3 unit, the structure of 5HF/AlF3 (3) bears a similar resemblance to the 4HF/AlF3 (1), whereas the structure of 5HF/AlF3 (4) is composed of the AlF3 fragment whose only two F atoms are involved in the connections to the HF molecules (alike it was observed for 2HF/AlF3 (1), 3HF/AlF3 (1), and 4HF/AlF3 (2)), however, the (HF)n internally H-bonded linkage consists of five HF molecules in the 5HF/AlF3 (4) case, see Fig. 1 and 2.
Finally, the lowest energy structure of 6HF/AlF3 (depicted as 6HF/AlF3 (1) in Fig. 1) is similar to the most stable isomer of 5HF/AlF3 (1). Namely, the planar AlF3 central fragment interacts with two (HF)3 internally H-bonded moieties, each of which forms (using its F-end) the dative F → Al(3p) bond and the hydrogen H⋯F–AlF2 bond (utilizing its H-terminus) with the AlF3 core. Again, viewing the 6HF/AlF3 (1) structure as composed of negatively charged AlF52− core, two H2F+ cations and two neutral HF molecules interacting with one another seems not justified. Instead, the interatomic distances and the results of the population analysis clearly indicate that this system represents the neutral AlF3 fragment interacting with six HF molecules (the mutual interactions among the HF systems are also present) and thus describing the resulting species by the 6HF/AlF3 formula is adequate. The gas phase acidity of 6HF/AlF3 (1), manifested by DE of 270 kcal mol−1, seems very similar to that predicted for the system having one HF molecule less (5HF/AlF3 (1)). Namely, the difference in DE for these two species does not exceed 1.5 kcal mol−1 (see Table 1). We view this result as very important because it shows that the addition of the sixth HF molecule to the 5HF/AlF3 system does not change its acidity, and thus the saturation of both DE and ΔGacid seems achieved (further discussion of the consequences of this result is provided in the closing section). We have also found three higher energy isomers of 6HF/AlF3 having their relative energies in the 6–19 kcal mol−1 range (with respect to the global minimum 6HF/AlF3 (1)), see structures 6HF/AlF3 (2, 3, and 4) depicted in Fig. 2. However, we believe that only one of them (6HF/AlF3 (2) whose structure resembles that of 5HF/AlF3 (3) with one more HF unit attached) might be considered as competitive with 6HF/AlF3 (1) because the relative energies of 6HF/AlF3 (3) and 6HF/AlF3 (4) seem too large (i.e., 9 and 19 kcal mol−1, respectively).
Having discussed the structures and acidities of the nHF/AlF3 (n = 1–6) systems (including their isomers possessing relative energies within 20 kcal mol−1), we present the lowest energy anionic structures of ((n − 1)HF/AlF4)− (n = 1–6) that are the resulting compounds of the nHF/AlF3 deprotonation. The equilibrium anionic structures are depicted in Fig. 3. One may notice that the AlF4 structural unit can be distinguished in all of these negatively charged systems, moreover, the population analysis and tetrahedral-like geometry of that AlF4 fragment indicate that the entire excess electron density is delocalized over its fluorine ligands. Thus, each ((n − 1)HF/AlF4)− anion (n = 1–6) is in fact composed of the quasi-tetrahedral AlF4− core and a certain number of HF molecules bound to its F ligands via the H-bonds. The symmetry of the resulting structures seems enforced by the number of the HF molecules attached, namely, the Td-symmetry corresponds to the AlF4− surrounded by either zero or four HF moieties, C3v-symmetry is achieved for either one or three HF molecules, whereas the C2v-symmetry anionic structure is observed when two HF systems are coordinated. Clearly, the presence of four electronegative ligands in the AlF4− anion indicates that this negatively charged species can be maximally stabilized by four HF molecules whereas the fifth hydrogen fluoride system remains outside this first coordination sphere (compare the structures of (4HF/AlF4)− and (5HF/AlF4)− anions in Fig. 3). Thus the lowest value of deprotonation energy (DE = 268.9 kcal mol−1, see Table 1) for the nHF/AlF3 superacids (n = 1–6) corresponds to the 5HF/AlF3 system while the effect of attaching an additional HF molecule is nearly marginal. Such an observation indicates the key role of microsolvation of an anionic species in the deprotonation process and its influence on the change in energy (with respect to the neutral microsolvated system).
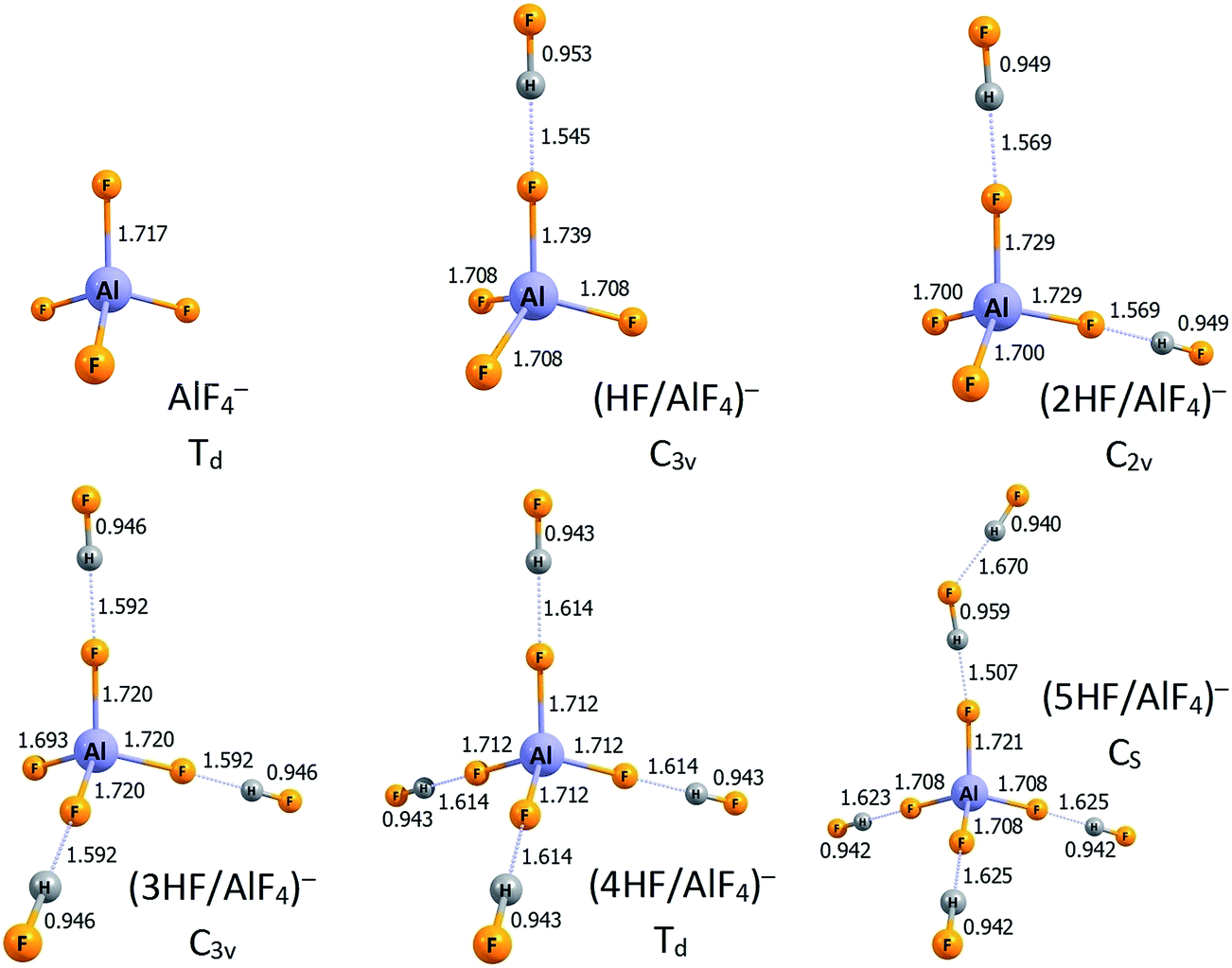 | ||
| Fig. 3 The structures of the lowest energy isomers of the ((n − 1)HF/AlF4)− anions (n = 1–6). Selected bond lengths are given in Å. Hydrogen bonds are represented by the dotted lines. | ||
3.2. The GeF4 Lewis acid surrounded by various number of HF molecules
The lowest energy structures of nHF/GeF4 (n = 1–6) systems and the corresponding higher energy isomers are depicted in Fig. 4 and 5, respectively. The structurally simplest HF/GeF4 system (i.e., the GeF4 Lewis acid interacting with one hydrogen fluoride molecule) has already been characterized as a promising superacid consisting of the HF fragment donating its F's lone pair to the empty germanium's 4p atomic orbital of the GeF4 quasi-tetrahedral unit.28 The DE of 295 kcal mol−1 and ΔGacid of 285 kcal mol−1 predicted for HF/GeF4 (see Table 2) are in good agreement with the earlier estimations obtained at a different theory level.28 The lowest energy structure of 2HF/GeF4 is assembled in a similar way, with one additional HF molecule attached, which results in the formation of two hydrogen bonds and in the shortening (by 0.22 Å) of the HF → Ge(4p) dative bond (in comparison to the HF/GeF4), see Fig. 4. The deprotonation energy estimated for the 2HF/GeF4 is smaller by 8 kcal mol−1 than that predicted for HF/GeF4, see Table 2, which indicates considerably stronger acidity of the former species. It seems important to notice that the DE drop observed for HF/GeF4 upon the second HF molecule addition is much larger than the analogous DE decrease noted for the HF/AlF3, see the preceding section. We also found another isomeric structure of 2HF/GeF4 (see 2HF/GeF4 (2) in Fig. 5) whose energy is larger by only 4 kcal mol−1 than the energy of global minimum 2HF/GeF4 (1). Similarly to the higher energy isomer of 2HF/AlF3 (2) (see Fig. 2), the structure of 2HF/GeF4 (2) might be described as two HF molecules attached to the opposite sides of the GeF4 central unit which allows for the formation of one dative bond (HF → Ge(4p)) and one hydrogen bond between HF and GeF4 fragments, see Fig. 5.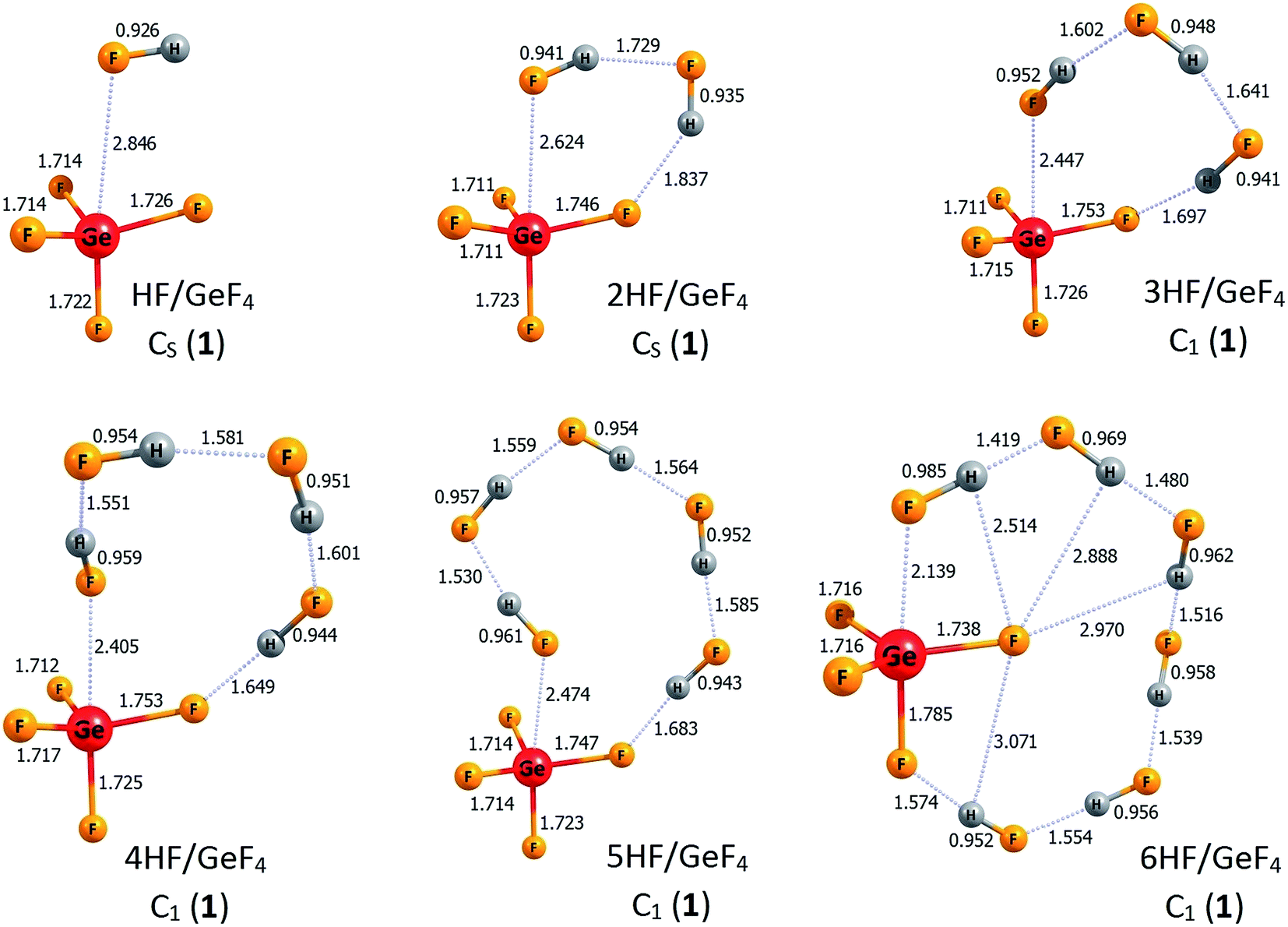 | ||
| Fig. 4 The structures of the lowest energy isomers of the nHF/GeF4 superacids (n = 1–6). Selected bond lengths are given in Å. Dative bonds and hydrogen bonds are represented by the dotted lines. | ||
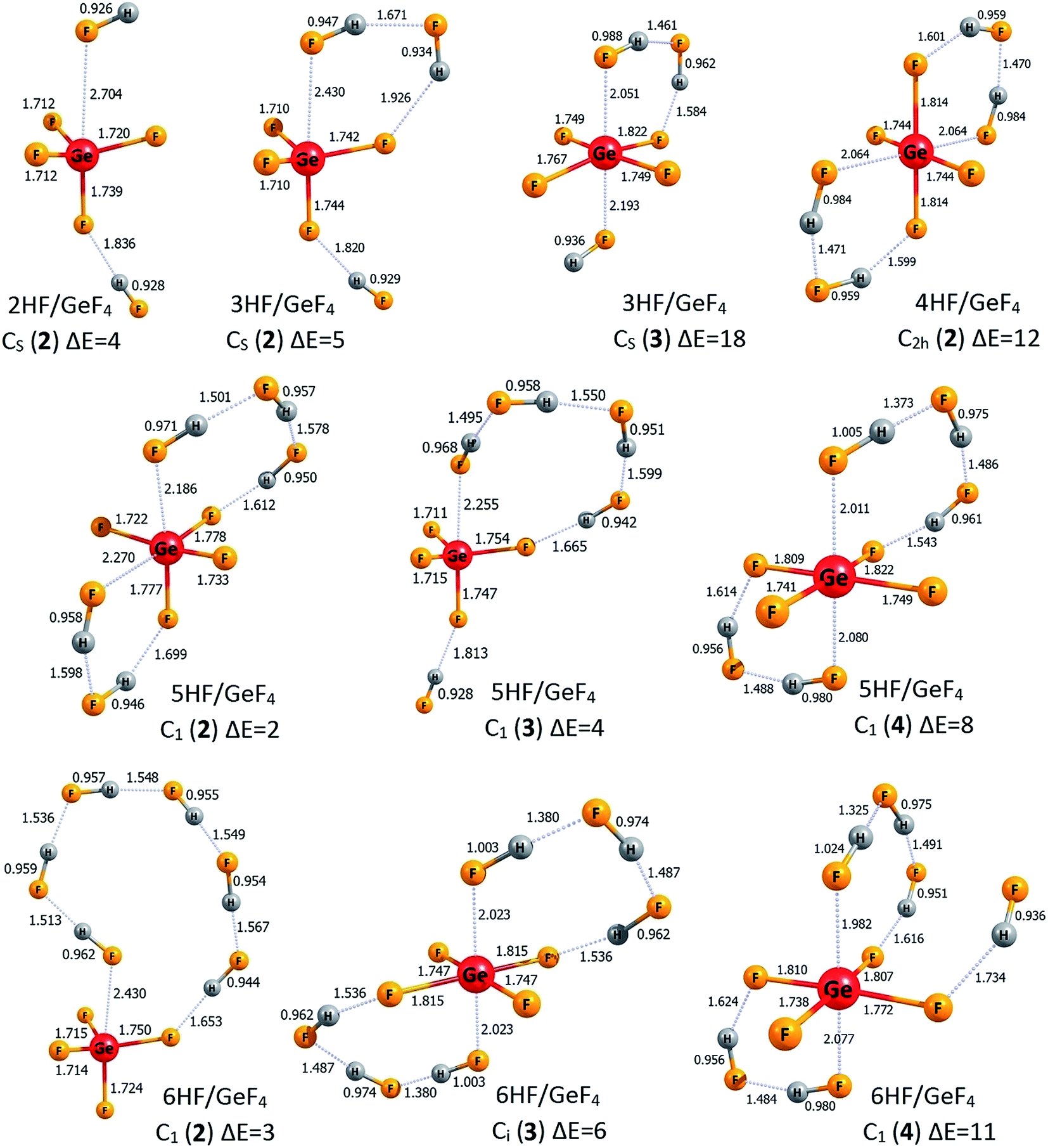 | ||
| Fig. 5 The structures of the higher energy isomers of the nHF/GeF4 superacids (n = 1–6). Selected bond lengths are given in Å. Relative energies (ΔE) with respect to the corresponding lowest energy isomers are given in kcal mol−1. Dative bonds and hydrogen bonds are represented by the dotted lines. | ||
| Species | Symmetry | DE | TΔS | ΔGacid |
|---|---|---|---|---|
| HF/GeF4 | C s | 294.8 | 4.1 | 285.0 |
| 2HF/GeF4 | C s | 286.9 | 7.4 | 273.7 |
| 3HF/GeF4 | C 1 | 281.4 | 8.5 | 267.2 |
| 4HF/GeF4 | C 1 | 278.1 | 9.7 | 262.7 |
| 5HF/GeF4 | C 1 | 276.1 | 10.8 | 259.8 |
| 6HF/GeF4 | C 1 | 277.6 | 13.6 | 259.5 |
The lowest energy structures of GeF4 interacting with three (3HF/GeF4 (1)), four (4HF/GeF4 (1)), five (5HF/GeF4 (1)), and six (6HF/GeF4 (1)) hydrogen fluoride molecules follow the same general pattern, see Fig. 4. Namely, in each of these systems, the internally H-bonded HF-chain ((HF)n) acts as a molecular “clasp” having two different ends – one of them (F-terminus) is involved in the (HF)n → Ge(4p) dative bond formation while the other (H-terminus) forms a hydrogen bond with one of the fluorine ligands the central tetrahedral-like GeF4 unit is decorated with. In the case of the 6HF/GeF4 (1), however, the (HF)6 clasp is long enough to attach its H-end to the fluorine ligand localized on the opposite side of the GeF4 fragment (with respect to the dative bond), see Fig. 4, which in turn allows for additional stabilization coming from the interaction of another F ligand with the hydrogen atoms of the (HF)6 chain. In each of the nHF/GeF4 (1) (n = 1–6) structures one may easily distinguish the central GeF4 unit having its four fluorine atoms localized in a quasi-tetrahedral manner around the Ge atom. On the other hand, it might be tempting to consider these structures as consisting of the GeF5 unit (forming a deformed trigonal bipyramid) – if such a view were applied, however, it would have required the presence of at least one H2F (likely cationic) moiety in the remaining fragment. As we verified (by analyzing the interatomic distances and partial atomic charges), such a treatment is not justified, mainly because all the H–F bond lengths are typical for HF molecules (0.93–0.99 Å), one F atom forms a significantly elongated bond with the Ge atom, and the partial atomic charges sum up to approximately zero for each HF fragment in the nHF/GeF4 (1) (n = 1–6) structures, see Fig. 4. Hence, we conclude that these lowest energy isomeric structures consist of the GeF4 (rather than GeF5 or GeF6) tetrahedral-like unit and the HF molecules attached, whereas the H2F+ fragments are absent.
The description of the nHF/GeF4 (n = 3–6) systems would not be complete if the higher energy isomeric structures were neglected. Hence, in Fig. 5 we present two additional isomers of 3HF/GeF4, one higher energy isomer of 4HF/GeF4, three isomeric structures of 5HF/GeF4, and three higher energy isomers of 6HF/GeF4. The relative energies of 3HF/GeF4 (2) and 3HF/GeF4 (3) (calculated as equal to 5 and 18 kcal mol−1, respectively) indicate that only the isomer 2 may compete with the 3HF/GeF4 (1) global minimum. The structure of 3HF/GeF4 (2) resembles that of 2HF/GeF4 (1) with the additional HF molecule attached to the opposite side of the GeF4, whereas the higher energy isomer 3HF/GeF4 (3) contains the quasi-planar GeF4 fragment (whose atypical structure is likely caused by the formation of two (instead of one) dative bonds), see Fig. 5. It seems also important to note that each of three HF molecules attached to this GeF4 fragment in 3HF/GeF4 (3) remains nearly intact. The attachment of four HF molecules to GeF4 leads to only one higher energy isomer (within 20 kcal mol−1) depicted in Fig. 5 (4HF/GeF4 (2)). The structure of 4HF/GeF4 (2) contains a quasi-planar GeF4 fragment forming two dative bonds and two hydrogen bonds with two (HF)2 clasps that link the ligands on the opposite sides, however, its energy is 12 kcal mol−1 higher than that of 4HF/GeF4 (1) and thus the formation of 4HF/GeF4 (2) isomer is not likely at low temperatures. The situation is different for the 5HF/GeF4 systems, namely, the relative energies of all three higher energy isomers (i.e., 5HF/GeF4 (2, 3, and 4)) are rather small (not exceeding 8 kcal mol−1) with respect to the global minimum 1, hence they may compete with the lowest energy isomer. The structures of 5HF/GeF4 consist of either a quasi-tetrahedral (2 and 3) or quasi-planar (4) GeF4 unit surrounded by five HF molecules in various ways, see Fig. 5. Finally, the relative energies of two (2 and 3) higher energy isomers of 6HF/GeF4 are small enough (3–6 kcal mol−1) to allow for their presence at low temperatures, while the energy of 6HF/GeF4 (4) was calculated to be 11 kcal mol−1 larger than that of 6HF/GeF4 (1). The structures of 6HF/GeF4 (3) and 6HF/GeF4 (4) contain a quasi-planar GeF4 fragment allowing for the formation of two dative bonds whereas the structure of 6HF/GeF4 (2) consists of a tetrahedral-like GeF4 moiety linked via one dative bond with the HF molecule localized nearby, see Fig. 5. Even though the structures of 6HF/GeF4 (3) and 6HF/GeF4 (4) might suggest the presence of the GeF62− fragment (which in turn would enforce the presence of two H2F+ fragments), we verified that such a supposition is not justified. As it was already discussed for the other structures, the interatomic distances in 6HF/GeF4 (3) and 6HF/GeF4 (4) indicate that all HF fragments resemble typical hydrogen fluoride molecules (with their H–F bond lengths spanning the 0.94–1.02 Å range) involved in the formation of various H-bonded structures rather than the cationic H2F+ fragments (such a conclusion is also additionally supported by the results of the population analysis showing approximately zero net charges on each HF molecule).
According to the nHF/AlF3 (n = 1–6) description provided in the preceding section, we briefly comment on the anionic structures of the nHF/GeF4 (n = 1–6) superacids (i.e., the corresponding anionic systems that result from deprotonation of those compounds). As for the ((n − 1)HF/AlF4)− anions (n = 1–6), we limit our discussion to the lowest energy anionic isomers ((n − 1)HF/GeF5)− anions (n = 1–6), while their excess electron detachment energies are not considered here (although we verified that all the presented anions are electronically stable systems). The lowest energy ((n − 1)HF/GeF5)− anionic structures are depicted in Fig. 6. In each case, the GeF5 structural unit can be distinguished with five F ligands forming a trigonal bipyramid around the germanium atom. The population analysis indicate that the GeF5 fragment holds the entire excess electron density in these anions (more precisely, the excess negative charge is delocalized over five fluorine ligands in GeF5). Thus, each of these negatively charged systems (i.e., ((n − 1)HF/GeF5)−) should be considered as the GeF5− anion with the (n − 1) hydrogen fluoride molecules attached. As depicted in Fig. 6, in all cases except (5HF/GeF5)−, the HF molecules are tethered to different fluorine ligands of GeF5−. Similarly to the ((n − 1)HF/AlF4)− species (n = 1–6) described in the preceding section, it turns out that the number of ligands in the GeF5− anion (whose presence as a central unit was confirmed in all ((n − 1)HF/GeF5)− cases) is crucial regarding its possible microsolvation by HF molecules. Namely, for the increasing value of n, the HF moieties are successively attached to different fluorine atoms that the GeF5− central unit consists of (with the only exception of (5HF/GeF5)− in which one F ligand is not involved, see Fig. 6). Again, this confirms the crucial role of microsolvation of an anionic species in the overall deprotonation process which manifests itself by the lowest values of deprotonation energy found for the 5HF/GeF4 and 6HF/GeF4 systems, see Table 2.
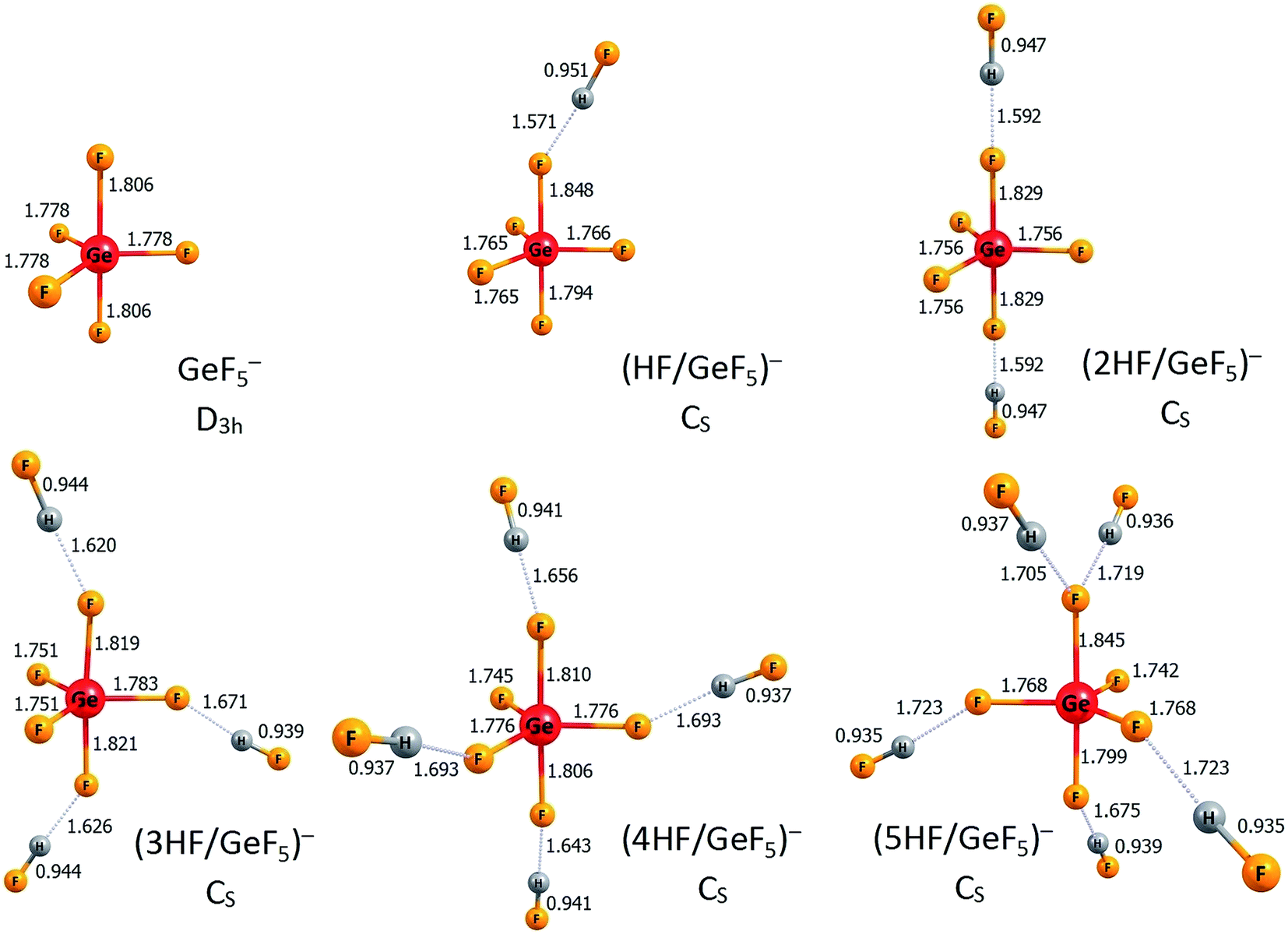 | ||
| Fig. 6 The structures of the lowest energy isomers of the ((n − 1)HF/GeF5)− anions (n = 1–6). Selected bond lengths are given in Å. Hydrogen bonds are represented by the dotted lines. | ||
As far as the DE and ΔGacid values of the nHF/GeF4 (n = 3–6) species are concerned, a gradual decrease of the deprotonation energy (accompanied by the Gibbs free deprotonation energy change) is observed when n develops from 3 to 5, see Table 2. The addition of the third HF molecule (resulting in the formation of 3HF/GeF4 (1)) leads to the DE = 281 kcal mol−1, while introducing fourth and fifth HF fragment lowers DE by another 3 and 2 kcal mol−1, respectively. Finally, we observe that the addition of the sixth hydrogen fluoride molecule has an opposite (although rather small) impact on the acidity, as it causes the deprotonation energy to increase by 1.5 kcal mol−1. As indicated in the preceding paragraph, the described DE (and thus ΔGacid) decrease is mainly caused by the ability of the central GeF5− unit to utilize its ligands to successively attach the HF molecules. The consequences of these findings are discussed in the following section.
3.3. Deprotonation energies of the nHF/AlF3 and nHF/GeF4
Although the energies of the deprotonation process for each investigated superacid were already briefly mentioned in the previous sections, we would like to summarize these results and formulate some important conclusions concerning the acidic strength. In general, the DE values calculated for the nHF/AlF3 and nHF/GeF4 (n = 1–6) gathered in Tables 1 and 2 indicate that the acidity strength increases with the increasing number (n) of HF molecules involved. It is manifested by the drop of the DE values (by 9 kcal mol−1 for nHF/AlF3 and by 19 kcal mol−1 for nHF/GeF4) with n developing from 1 to 5. In both cases, the attachment of the sixth HF molecule does not lead to the further acidity increase. The estimated deprotonation energies (depicted in Fig. 7) seem to converge to some limiting values of ca. 269–270 kcal mol−1 (for nHF/AlF3) and 276–277 kcal mol−1 (for nHF/GeF4), see Tables 1 and 2. These deprotonation energy changes are accompanied by the similar ΔGacid changes whose values converge to 251 kcal mol−1 (for 6HF/AlF3) and 260 kcal mol−1 (for 6HF/GeF4).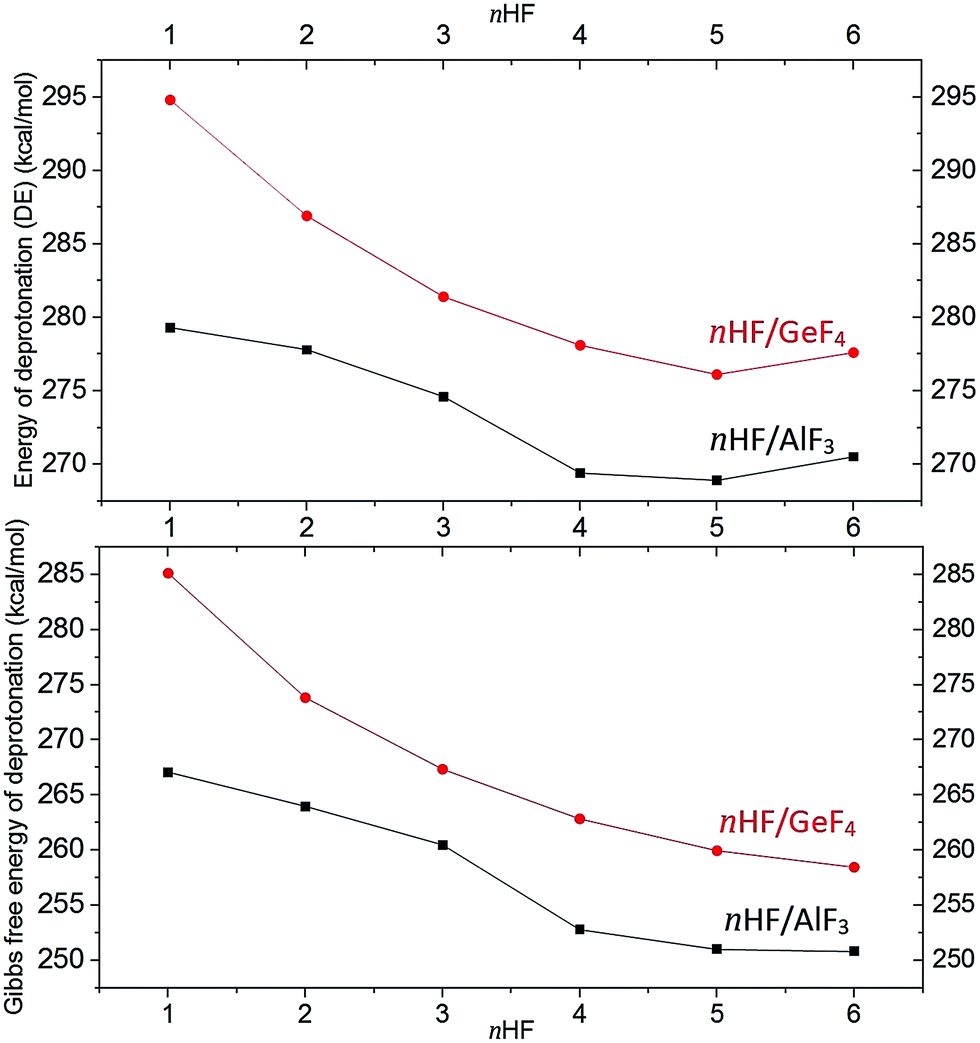 | ||
| Fig. 7 Deprotonation energies (top) and the Gibbs free deprotonation energies (bottom) (in kcal mol−1) for nHF/AlF3 (black squares) and nHF/GeF4 (red circles) superacids (n = 1–6). | ||
The results described above suggest that the successive attachment of HF molecules to either AlF3 or GeF4 results in a gradual increase of the acidity strength. This acidity strength increase is less and less visible for growing number of HF molecules which leads to the saturation of the DE and ΔGacid values. The saturation of deprotonation energy for both nHF/AlF3 and nHF/GeF4 superacids is likely caused by the limited ability of the AlF4− and GeF5− anions to attach HF molecules while forming their first coordination spheres. Taking into account that the number of HF molecules introduced (i.e., up to 6) is larger than the number of electronegative sites in either AlF4− and GeF5− anion, we believe that such a saturation is achieved for both the nHF/AlF3 and nHF/GeF4 series (n = 1–6). Certainly, the slower DE saturation process for nHF/GeF4 than for nHF/AlF3 is caused by the larger number of accessible fluorine ligands in GeF5− where the surrounding HF molecules can be attached to. Hence, we speculate that our final limiting DE and ΔGacid values (DE = 269–270 kcal mol−1, ΔGacid = 251 kcal mol−1 for nHF/AlF3 and DE = 276–277 kcal mol−1, ΔGacid = 260 kcal mol−1 for nHF/GeF4) are reasonable and reliable approximations of the true gas phase deprotonation energies of these superacids (assuming that such superacids are prepared by combining either AlF3 or GeF4 Lewis acid with the excess of anhydrous hydrogen fluoride).
4. Conclusions
On the basis of our quantum chemical calculations performed at the QCISD/6-311++G(d,p)//B3LYP/6-311++G(d,p) level for the nHF/AlF3 and nHF/GeF4 (n = 1–6) neutral superacids (i.e., AlF3 and GeF4 Lewis acids surrounded by n hydrogen fluoride (Brønsted acid) molecules) and their corresponding anions (i.e., negatively charged systems formed by deprotonation of the nHF/AlF3 and nHF/GeF4) we formulated the following conclusions:(i) The acidic strength of the Brønsted/Lewis superacids (approximated here by the deprotonation energies, DE) prepared by combining the Lewis acid with the excess of Brønsted acid should not be estimated by assuming a single Lewis acid molecule interacting with one Brønsted acid molecule as a building block.
(ii) Each Lewis acid moiety is capable of interacting (dative bond and hydrogen bond formation) with more than one Brønsted acid molecule, as verified for AlF3 and GeF4 Lewis acids surrounded by various number (from 1 to 6) hydrogen fluoride molecules.
(iii) The successive attachment of HF molecules to either AlF3 or GeF4 gradually increases their acidity strength (manifested by the DE decrease) which leads to the saturation of the DE value achieved for the nHF/AlF3 and nHF/GeF4 superacids for n = 5–6.
(iv) The microsolvation of an anionic species generated in the course of a deprotonation reaction (i.e., either AlF4− or GeF5−) plays a key role in the process as it influences the change in energy with respect to the neutral microsolvated species.
(v) The final values of the deprotonation energies estimated here for the Brønsted/Lewis superacids prepared by combining either AlF3 or GeF4 Lewis acids with the excess of anhydrous hydrogen fluoride are equal to 269–270 kcal mol−1 and 276–277 kcal mol−1, respectively.
Although our conclusions were formulated by investigating only two arbitrarily chosen Brønsted/Lewis superacids we believe they should be more general in a sense that they might be extended to cover all such superacids prepared in the similar way. Thus the future theoretical predictions of the gas phase acidity of Brønsted/Lewis superacids cannot be limited to evaluating the properties of a building block assumed as a single Lewis acid molecule interacting with one Brønsted acid molecule.
Acknowledgements
This research was supported by the Polish Ministry of Science and Higher Education grant No. DS 530-8376-D499-15 and by the grant no. PSPB-051/2010 from Switzerland through the Swiss Contribution to the enlarged European Union (to P. S.). The calculations have been carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (http://wcss.pl) grant No. 350.References
- R. J. Gillespie and T. E. Peel, Adv. Phys. Org. Chem., 1971, 9, 1–24 CrossRef CAS.
- R. J. Gillespie and T. E. Peel, J. Am. Chem. Soc., 1973, 95, 5173–5178 CrossRef CAS.
- N. F. Hall and J. B. Conant, J. Am. Chem. Soc., 1927, 49, 3047–3061 CrossRef CAS.
- G. A. Olah and J. Lukas, J. Am. Chem. Soc., 1967, 89, 2227–2228 CrossRef CAS.
- A. F. Bickel, C. J. Gaasbeek, H. Hogeveen, J. M. Oelderik and J. C. Platteeuw, J. Chem. Soc., Chem. Commun., 1967, 13, 634–635 RSC.
- H. Hogeveen and A. F. Bickel, J. Chem. Soc., Chem. Commun., 1967, 13, 635–636 RSC.
- G. A. Olah and R. H. Schlosberg, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS.
- H. Hogeveen and A. F. Bickel, Recl. Trav. Chim. Pays-Bas, 1969, 88, 371–378 CrossRef CAS.
- G. A. Olah, Y. Halpern, J. Shen and Y. K. Mo, J. Am. Chem. Soc., 1971, 93, 1251–1256 CrossRef CAS.
- A. H. Otto, T. Steiger and S. Schrader, Chem. Commun., 1998, 3, 391–392 RSC.
- I. A. Koppel, P. Burk, I. Koppel, I. Leito, T. Sonoda and M. Mishima, J. Am. Chem. Soc., 2000, 122, 5114–5124 CrossRef CAS.
- K. E. Gutowski and D. A. Dixon, J. Phys. Chem. A, 2006, 110, 12044–12054 CrossRef CAS PubMed.
- A. K. Srivastava and N. Misra, Polyhedron, 2015, 102, 711–714 CrossRef CAS.
- J. Axhausen, C. Ritter, K. Lux and A. Kornath, Z. Anorg. Allg. Chem., 2013, 639, 65–72 CrossRef CAS.
- C. Bour, R. Guillot and V. Gandon, Chem.–Eur. J., 2015, 21, 6066–6069 CrossRef CAS PubMed.
- G. A. Olah, G. K. Prakash and J. Sommer, Science, 1979, 206, 13–20 CAS.
- D. Touiti, R. Jost and J. Sommer, J. Chem. Soc., Perkin Trans., 1986, 2, 1793–1797 RSC.
- R. Jost and J. Sommer, Rev. Chem. Intermed., 1988, 9, 171–199 CrossRef CAS.
- J. Axhausen, K. Lux and A. Kornath, Angew. Chem., Int. Ed. Engl., 2014, 53, 3720–3721 CrossRef CAS PubMed.
- C. Sikorska, S. Freza and P. Skurski, J. Phys. Chem. A, 2010, 114, 2235–2239 CrossRef CAS PubMed.
- M. Czapla and P. Skurski, Chem. Phys. Lett., 2015, 630, 1–5 CrossRef CAS.
- M. Czapla and P. Skurski, Phys. Chem. Chem. Phys., 2015, 17, 19194–19201 RSC.
- S. Senger and L. Radom, J. Phys. Chem. A, 2000, 104, 7375–7385 CrossRef CAS.
- G. Zhong, B. Chan and L. Radom, Org. Lett., 2009, 11, 749–751 CrossRef CAS PubMed.
- M. Czapla and P. Skurski, J. Phys. Chem. A, 2015, 119, 12868–12875 CrossRef CAS PubMed.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- X.-B. Wang, C.-F. Ding, L.-S. Wang, A. I. Boldyrev and J. Simons, J. Chem. Phys., 1999, 110, 4763–4771 CrossRef CAS.
- M. Czapla, I. Anusiewicz and P. Skurski, Chem. Phys., 2016, 465–466, 46–51 CrossRef CAS.
- F. A. Hohorst, L. Stein and E. Gebert, Inorg. Chem., 1975, 14, 2233–2236 CrossRef CAS.
- T. Drews, W. Koch and K. Seppelt, J. Am. Chem. Soc., 1999, 121, 4379–4384 CrossRef CAS.
- K. O. Christe, C. J. Schack and R. D. Wilson, Inorg. Chem., 1977, 16, 849–854 CrossRef CAS.
- J. E. Roberts and A. W. Laubengayer, J. Am. Chem. Soc., 1957, 79, 5895–5897 CrossRef CAS.
- K. O. Christe, D. A. Dixon, D. J. Grant, R. Haiges, F. S. Tham, A. Vij, V. Vij, T.-H. Wang and W. W. Wilson, Inorg. Chem., 2010, 49, 6823–6833 CrossRef CAS PubMed.
- M. D. Lind and K. O. Christe, Inorg. Chem., 1972, 11, 608–612 CrossRef CAS.
- J. F. Lehmann, G. J. Schrobilgen, K. O. Christe, A. Kornath and R. J. Suontamo, Inorg. Chem., 2004, 43, 6905–6921 CrossRef CAS PubMed.
- D. E. McKee, C. J. Adams and N. Bartlett, Inorg. Chem., 1973, 12, 1722–1725 CrossRef CAS.
- Z. Mazej and E. Goreshnik, Inorg. Chem., 2008, 47, 4209–4214 CrossRef CAS PubMed.
- H. S. A. Elliott, J. F. Lehmann, H. P. A. Mercier, H. D. B. Jenkins and G. J. Schrobilgen, Inorg. Chem., 2010, 49, 8504–8523 CrossRef CAS PubMed.
- J. F. Lehmann, D. A. Dixon and G. J. Schrobilgen, Inorg. Chem., 2001, 40, 3002–3017 CrossRef CAS PubMed.
- J. Bacon, P. A. W. Dean and R. J. Gillespie, Can. J. Chem., 1970, 48, 3413–3424 CrossRef CAS.
- W. W. Wilson, R. C. Thompson and F. Aubke, Inorg. Chem., 1980, 19, 1489–1493 CrossRef CAS.
- R. Faggiani, D. K. Kennepohl, C. J. L. Lock and G. J. Schrobilgen, Inorg. Chem., 1986, 25, 563–571 CrossRef CAS.
- R. J. Gillespie and K. C. Moss, J. Chem. Soc. A, 1966, 1170–1175 RSC.
- B. Bonnet and G. Mascherpa, Inorg. Chem., 1980, 19, 785–788 CrossRef CAS.
- J. C. Culmann, M. Fauconet, R. Jost and J. Sommer, New J. Chem., 1999, 23, 863–867 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS.
- J. Gauss and D. Cremer, Chem. Phys. Lett., 1988, 150, 280–286 CrossRef CAS.
- E. A. Salter, G. W. Trucks and R. J. Bartlett, J. Chem. Phys., 1989, 90, 1752–1766 CrossRef.
- B. H. Besler, K. M. Merz Jr and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The Cartesian coordinates of all the structures presented in this work and their corresponding energies are included. See DOI: 10.1039/c6ra02199a |
| This journal is © The Royal Society of Chemistry 2016 |