
Binder-free lithium ion battery electrodes made of silicon and pyrolized lignin†
Abstract
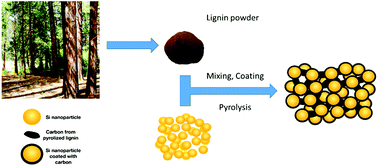
The synthesis, characterization, and performance of a binder-free negative electrode for a lithium-ion battery, consisting of renewable biopolymer lignin and silicon nanoparticles, are reported. By mixing, coating, and subsequent pyrolization, we fabricated uniformly interconnected core–shell composite films of Si/C directly on the current collector, allowing for the assembly of coin-cells without the need of binder and conductive carbon. An excellent electrochemical performance was observed with a high specific capacity of 1557 mA h g−1 and a stable rate performance from 0.18 A g−1 to 1.44 A g−1. Moreover, the Si–pLig electrode can be reversibly cycled at 0.54 A g−1 with 89.3% capacity retention over 100 cycles. We also unveil a beneficial effect of 0.5% polyethylene oxide (PEO) on the morphology and electrochemical behavior of the Si/C composite electrodes.
 Please wait while we load your content...
Please wait while we load your content...

