
A sensitive electrochemiluminescent immunosensor based on 3D-flower-like MoS2 microspheres and using AuPt nanoparticles for signal amplification
 a
and
a
and
Abstract
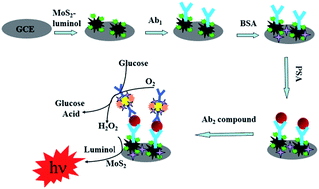
We herein report the synthesis of MoS2, consisting of three-dimensional flower-like microspheres assembled by bent flakes with a thickness of several nanometers. The diameter of theses spheres was approximately 1.5 μm, and they showed good catalysis of H2O2 and could increase the electrochemiluminescence (ECL) intensity of luminol. The ECL properties of luminol were investigated and a type of luminol composite (MoS2–luminol) was prepared. Glucose oxidase–AuPt nanoparticles (GOx–AuPt) showed excellent catalytic performance for the reduction reaction of glucose. The prepared GOx–AuPt and MoS2–luminol were applied in a sandwich-type ECL immunosensor for prostate-specific antigen (PSA). In the immunosensor, MoS2–luminol acted as a solid support for PSA primary antibody, and GOx–AuPt was employed as a support for PSA secondary antibody. With the addition of glucose, hydrogen peroxide was prepared and the ECL properties of the MoS2–luminol were enhanced. The proposed immunosensor enabled PSA concentrations to be determined in the range of 0.001 ng mL−1 to 100 ng mL−1, with a detection limit of 0.28 pg mL−1. The experimental results indicated that the immunosensor exhibited simple instrumentation, high sensitivity, wide linear range and excellent analytical performance, and could be a promising technique for tumor marker detection.
 Please wait while we load your content...
Please wait while we load your content...

