
The synthesis of chiral triphenylpyrrole derivatives and their aggregation-induced emission enhancement, aggregation-induced circular dichroism and helical self-assembly†
Abstract
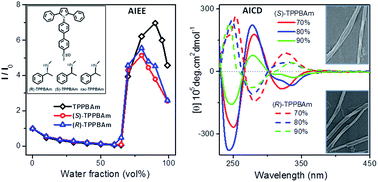
A pair of enantiomers ((R)-TPPBAm and (S)-TPPBAm) and their raceme (rac-TPPBAm) were designed and prepared by conjugating (R)-, (S)- or racemic 1-phenylethylamine to an aggregation-induced emission enhancement (AIEE) active triphenylpyrrole fluorophore. The three target compounds were thoroughly characterized and their optical properties were systematically investigated. The fluorescence analyses indicate that they all retain the AIEE activities originating from the triphenylpyrrole moiety, irrespective of the attaching groups. More importantly, both the enantiomers containing (R)- or (S)-1-phenylethylamine attachment exhibit aggregation-induced circular dichroism (AICD) features with mirror-image signals. Besides, they also exhibit circularly polarized luminescence (CPL) with an emission dissymmetry factor (gem) from 1.5 × 10−4 to 3 × 10−3 for (R)-TPPBAm and −1.3 × 10−4 to −4 × 10−3 for (S)-TPPBAm in aggregate states. As expected, consistent with the variations of their CD signals, (R)-TPPBAm and (S)-TPPBAm could self-assemble into helical nanofibers with the opposite screw direction during the aggregation process in the THF–water mixed solution, while the racemic compound rac-TPPBAm exhibits no CD and CPL signals, and self-assembles to form nanoparticles blocks. These results demonstrate that the morphologies and optical activities can be controlled simultaneously without losing the solid-state emission performance of the material by attaching a chiral group to an AIEE fluorophore, which could shed light on the design of optical active fluorophores for sensitive and time-efficient enantiomer determination.
 Please wait while we load your content...
Please wait while we load your content...

