
Application of magnetic lamotrigine-imprinted polymer nanoparticles as an electrochemical sensor for trace determination of lamotrigine in biological samples†
Abstract
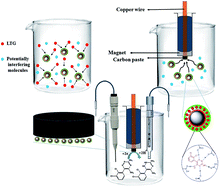
In the present work, a differently shaped electrode based on carbon paste was designed using a magnet at the back of a carbon paste (made a magnet containing a carbon paste electrode) for determination of lamotrigine, using magnetic molecularly imprinted polymer (Magnetic-MIP) nanoparticles. Magnetic-MIPs have been synthesized for the preconcentration and selective trace determination of lamotrigine (LTG) in urine and plasma samples. The obtained sorbent was characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM), infrared absorption spectroscopy (IR) and thermogravimetric analysis (TG). Separation of the sorbent from the sample solution was simply achieved by an electrode via a magnetic field from the magnet containing electrode. Determination of the extracted drug was performed by the differential pulse voltammetry (DPV) technique. Under the optimum extraction conditions, the method detection limits were 4.7 pM (based on a S/N of 3) for urine samples and 5.9 pM for plasma samples. Two linear dynamic ranges from 0.01–1.0 nM and 1.0–200 nM were obtained for LTG in biological samples.
 Please wait while we load your content...
Please wait while we load your content...

