
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stable organic–inorganic junction composed of hydrogenated titania nanotubes infiltrated by a conducting polymer†
Katarzyna Siuzdak*a,
Mariusz Szkodab,
Anna Lisowska-Oleksiakb,
Jakub Karczewskic and
Jacek Ryld
aCentre, for Plasma and Laser Engineering, Szewalski Institute of Fluid Flow Machinery, Polish Academy of Science, Fiszera 14, Gdańsk 80-231, Poland. E-mail: ksiuzdak@imp.gda.pl; Fax: +48 58 3416144; Tel: +48 58 6995294
bDepartment of Chemistry and Technology of Functional Materials, Chemical Faculty, Gdańsk University of Technology, Narutowicza 11/12, Gdańsk 80-233, Poland
cFaculty of Applied Physics and Mathematics, Gdańsk University of Technology, Narutowicza 11/12, 80-233 Gdańsk, Poland
dDepartment of Electrochemistry, Corrosion and Materials Engineering, Gdańsk University of Technology, Narutowicza 11/12, 80-233 Gdańsk, Poland
First published on 21st March 2016
Abstract
A poly(3,4-ethylenedioxythiophene) conducting polymer doped with poly(2-styrene sulfonate) (pEDOT:PSS) was efficiently electrodeposited on a layer composed of ordered titania nanotubes. TiO2 nanotubes were formed during an anodization process and, after calcinations, a layer was subjected to hydrogen plasma. Hydrogenation leads to Ti(III) formation, a decrease in resistance, and a huge increase of donor density when compared with pure titania. According to a detailed structure analysis, the coverage by the polymer matrix is uniform on the entire titania surface as well as along the tubes. The composite material exhibits highly enhanced anodic photocurrent (106 μA cm−2) when compared with hydrogenated titania H–TiO2 (54 μA cm−2) or pure polymer film (2 μA cm−2). Moreover, H–TiO2/pEDOT:PSS is characterized with high photostability displayed during prolonged illumination. The proposed hydrogenation approach could be regarded as a facile titania modification for further electrochemical modifications.
Introduction
Research into photoactive materials has long aimed at identifying low-cost semiconducting materials and facile preparation of photovoltaic junctions. Among many inorganic semiconductors, titanium dioxide, in particular well-ordered TiO2 nanotubes (TiO2NTs), has attracted a great deal of attention because of its advantages of an efficient pathway for vectorial electron transfer, light propagation through the nanoarchitecture, and a large surface to volume area.1,2 The ordered titania nanotubes are formed via electrochemical anodization of Ti plate immersed in fluoride-rich electrolyte. Precise optimization of anodization parameters, such as voltage, process duration, current distribution, temperature, and electrolyte composition, allows control of the morphology of the obtained TiO2NT film.3TiO2 nanotubes have replaced TiO2 nanoparticles in many applications because the anodization procedure for the nanotubes is relatively simple and can be highly controlled. As an n-type semiconductor, TiO2NTs are widely used in sensors, electrochromic devices,4 and devices for energy conversion and storage.5 In the case of photoconversion devices, titanium dioxide is widely used in dye-sensitized solar cells, inverted organic solar cells as a buffer layer,6 and blended with organic sensitizers or donors.7 In the TiO2/polymer-based solar cell, TiO2 forms p–n heterojunctions in which the conducting polymer acts as a p-element. Highly ordered heterojunctions obtained by filling metal oxide nanostructure with conducting polymer have distinctive advantages over composites in which nanoparticles are randomly distributed inside polymer because of a more direct and more efficient charge transfer path.8 Therefore, there has been intense research on TiO2NT/conducting polymer heterojunctions and their applications for photocurrent generation,9 water splitting,10 and energy storage.11,12 Polyaniline,13 polypyrrole,14 or poly(3,4-ethylenedioxythiophene)15 are used as conducting polymers. In most cases, polymer is formed inside the TiO2NTs via electrochemical16 or photoelectrochemical polymerization.17 Depending on the polymerization conditions (potentiostatic, potentiodynamic) and titania morphology, polymer matrix can be introduced inside the tubes or deposited on the outer wall.18 Furthermore, there have also been some attempts to facilitate and make deposition of polymer more uniform. Mainly, these involve titania modification by silane derivatives19 or electrode illumination during the polymerization process16 (photoelectrochemical polymerization). However, titania nanotubes obtained by anodization have fairly high resistance, which hampers direct electrodeposition of conducting polymer, and, additionally, an insulating barrier layer is present between titanium metal plate and tubular nanostructure.20 Recently, hydrogenation treatment was proposed to achieve improved conductivity of TiO2, and ultimately to achieve effective polymer deposition.21 According to the literature reports, TiO2 treatment in hydrogen atmosphere enhances electrochemical activity and conductivity because of increased donor density and introduction of surface hydroxyl groups.22,23 Thus, such an approach could lead to activation of the titania surface to allow for further electrochemical modifications.
This paper focuses on formation of composite material containing ordered titania nanotubes infiltrated with conducting polymer: pEDOT:PSS. Titanium nanotubes were formed on titanium foil via anodization in fluoride-rich electrolyte, followed by calcination to obtain the crystalline phase of the materials. Anatase NTs were subjected to hydrogen plasma to activate the surface for further polymer deposition. Polymer was introduced inside the nanostructure by potentiostatic electropolymerization with Ti/TiO2NTs acting as a working electrode immersed in monomer and counter ion solution. The morphology, crystal structure, and composition were investigated using scanning electron microscopy, Raman, UV-Vis, and X-ray photoelectron spectroscopy techniques. Electrochemical activity of the composite electrode, as well as pure and hydrogenated titania, was tested by means of cyclic voltammetry and electrochemical impedance spectroscopy measurements. To verify the stability of the TiO2NTs/polymer junction under illumination, long transient photocurrent measurements were performed.
Experimental
Preparation of hydrogenated TiO2
The TiO2NT samples were prepared via two-step anodization according to a previously optimized procedure.24 Before anodization, the substrate sample (Ti plate, Steam, 99.7%) was ultrasonically cleaned, with acetone, ethanol, and water, in that order, for 10 minutes in each solvent, and ultimately dried in air. The process was realized in a two-electrode configuration at room temperature, in which the titanium plate served as an anode and the platinum mesh as a cathode with a fixed distance of 2 cm between them. The first anodization took place in electrolyte containing ethylene glycol (EG), 0.27 M NH4F, and 1 vol% of deionized water. Anodization was performed at 40 V for 2 h, with an initial voltage rate of 0.1 V s−1, then the titanium plate was immersed for 12 h in 0.5% wt solution of oxalic acid to remove the rugged nanotube layer. The second anodization process was performed on the as-cleaned titanium plate under the same conditions as those during the first anodization. To remove surface debris, the titanium plates covered with nanotubes were immersed in 0.05% wt HF for 180 s. After the second anodization, the samples were rinsed with deionized water, dried in air, and thermally treated at 450 °C for 2 h with a heating rate of 2 °C min−1 to transform the amorphous TiO2 into a crystalline phase.Finally, the TiO2NTs were subjected to a hydrogen environment in a plasma reactor (Electronic Diener) operating at a power of 40 W, at room temperature, for 60 min. The obtained sample was coded as H–TiO2NTs.
Fabrication of H–TiO2NTs/PEDOT-PSS
Electrochemical polymerization was carried out to prepare H–TiO2NTs/PEDOT:PSS. After a series of optimization experiments, electrochemical deposition was performed in an aqueous solution containing 0.1 M NaPSS and 0.001 M EDOT, by potentiostatic polymerization of 1.6 V vs. Ag/AgCl/0.1 M KCl consuming charge of 30 mC cm−2. The hydrogenated titania sample constituted a working electrode. The reference electrode was Ag/AgCl/0.1 M KCl, and a platinum mesh was used as a counter electrode. Finally, the working electrode was washed with deionized water and dried in air.Characterization
The surface morphology and cross-section were examined using the Schottky field emission scanning electron microscope (FEI Quanta FEG 250) with an ET secondary electron detector. The beam accelerating voltage was kept at 10 kV. For elemental analysis, energy dispersive X-ray spectroscopy (EDX) was performed using an EDAX Genesis APEX 2i with ApolloX SDD spectrometer on a particular area of each sample. To confirm uniform polymer deposition, elemental analysis was completed on the surface and across the composite layer. The chemical nature of elements and binding properties of the surface were studied by X-ray photoelectron spectroscopy (XPS), using the Escalab 250Xi from ThermoFisher Scientific. High-resolution spectra were recorded at an energy step size of 0.1 eV at a pass energy of 10 eV. To normalize the spectroscopic measurements, the X axis (binding energy, Ebin) of the XPS spectrum was calibrated for peak characteristics of neutral carbon 1s (Ebin = 284.6 eV). UV-Vis reflectance spectra of titania nanotubes were measured with a dual beam UV-Vis spectrophotometer (Lambda 35, PerkinElmer) equipped with a diffuse reflectance accessory. The spectra were registered in a range of 300–700 nm, with a scanning speed of 60 nm min−1. Bandgap energy values were determined as the intercept of the tangent of the plot of transformation of the Kubelka–Munk function. This method was used to determine the bandgap energy for pure and H–TiO2NTs. Raman spectra were recorded using a confocal micro-Raman spectrometer (InVia, Renishaw) with sample excitation, by means of an argon ion laser emitting at 514 nm operating at 5% of its total power (50 mW).Electrochemical and photoelectrochemical measurements
Electrochemical measurements were carried out using an AutoLab PGStat 302N potentiostat-galvanostat system (Methrom, Autolab) in the standard three-electrode assembly, with titanium foil covered by nanotubes serving as the working electrode (active surface area of 0.3 cm2). Electrodes were tested in contact with deaerated aqueous 0.5 M K2SO4 electrolyte. An electrochemical cell was equipped with a quartz window. The photoactive layers were illuminated with a 150 W Xenon lamp (Osram XBO 150) and an AM 1.5 filter was used to obtain simulated solar light. An automated light chopper with a period of 90 s was used as a light source. The irradiance of incident light was measured to be 100 mW cm−2 using a reference cell (Si solar cell, Rera). The photocurrent measurements were carried out at + 0.5 V vs. Ag/AgCl/0.1 M KCl bias voltage. To perform measurements under the visible light range, an additional cut-off optical filter (λ > 420 nm) was added to the experimental setup.Electrochemical impedance spectroscopy (EIS) measurements for titania as well as composite samples were conducted at a frequency range between 20 kHz and 0.1 Hz, covering 75 points and with 10 mV amplitude of the AC signal. To perform the Mott–Schottky analysis, impedance spectra were recorded at different potentials in the range between +0.9 V and −0.6 V vs. Ag/AgCl/0.1 M KCl. Before each spectrum registration, the potential was held to achieve a steady state condition. The impedance data were analyzed on the basis of an electric equivalent circuit (EEQC) using an EIS spectrum analyzer program.
Results and discussion
Characterization
Scanning electron microscopy was employed to investigate the surface morphology of H–TiO2 NT and composite H–TiO2NTs/pEDOT:PSS. Fig. 1a and b shows the top-view and cross-section SEM images of pure and H–TiO2NTs layer, respectively. The obtained nanotubes were ordered and aligned. When comparing the SEM images registered for both materials, no difference was observed in the nano-architecture between the hydrogenated and pure titania. This suggests clearly that the applied plasma treatment did not influence the morphology, and only potential changes in the chemical states could have an impact on the electrochemical properties of titania. High resolution images allowed determination of tube dimensions: internal diameter of 100 nm, a tube-wall thickness of 25 nm, and tube length of 2.5 μm. Fig. 1c shows the layer of titania nanotubes infiltrated with pEDOT:PSS. The images demonstrate the growth of polymer film on the surface as well as along the tubes. In some cases, the holes are entirely filled with pEDOT:PSS matrix. This type of layer growth allows for close metal oxide–polymer contact, which is required in p–n heterojunction photovoltaic cells. | ||
| Fig. 1 SEM images of (a) pure TiO2, (b) hydrogenated TiO2, (c) H–TiO2NTs/pEDOT:PSS composite material, (d) remaining pEDOT:PSS after H–TiO2 removal. | ||
To determine the location of deposited polymer, the obtained composite H–TiO2NTs/pEDOT:PSS was immersed in diluted hydrofluoric acid (0.5 M), which selectively dissolves TiO2 nanotubes.25 The efficiency of crystalline titania form removal was highlighted by the Raman spectrum in which no bands typical for anatase were registered. After removal of the titania network acting as polymer scaffolding, pEDOT:PSS collapses and does not retain the ordered structure (see, Fig. 1d). The observed shape of remaining polymer allows us to conclude that polymerization takes place both inside and in-between nanotubes, as previously observed by Janáky et al.26
Energy dispersive X-ray spectroscopy was used to study distribution of the polymer matrix over the titania surface and along the tubes. Fig. S1a–c† shows clearly that the distribution of elements (C, O, S) included in pEDOT:PSS is homogeneous. Therefore, the material composition was preserved when the sample was investigated at its surface. The atomic content of each element present in the composite sample is as follows: Ti: 33.04 at%; O: 51.23 at%, C: 15.01 at%, S: 0.36 at%. As it is known that the polymer also contains oxygen atoms, the titanium to oxygen ratio deviates from TiO2 stoichiometry suggesting an oxygen deficit in the inorganic semiconductor. Furthermore, local EDX analysis performed at nine different points across composite materials (see Fig. S2†) showed uniform distribution of sulfur that builds pEDOT:PSS, and oxygen and carbon species present in the polymer and titania material. Thus, it is suggested that polymer penetrates the free space in the titania nanostructure from the surface region down to the tube base. However, as EDX inspection is characterized by limited resolution, further investigations using XPS were carried out under the etching mode allowing more valuable analysis.
The Raman spectra recorded for composite material and its pure counterparts, hydrogenated titania nanotubes and conducting polymer, are shown in Fig. 2. Raman spectra for unmodified titania nanotubes are given as a reference. A number of bands characteristic of the pure anatase crystalline form of TiO2 were identified in samples of pristine titania and composite material. The bands located at 144, 198, 395, 516, and 637 cm−1 are attributed to Eg(1), Eg(2), B1g, A1g, and Eg(3) active anatase modes, respectively.27,28 Characteristic bands of pEDOT:PSS are observed in pure polymer and composite material. The main band at 1433 cm−1 can be attributed to symmetric (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)–O bond vibrations in the thiophene ring. Two peaks at 1495 cm−1 and 1561 cm−1 could be interpreted as asymmetric CC modes in polymer chains.29 The peaks 1366 and 1267 cm−1 are characteristic of vibrations of single-bonded carbon atoms in the polymer chain and in the thiophene ring, respectively. The oxyethylene ring deformations can be identified at 988, 571, and 442 cm−1.30 The Raman spectra registered for the pure titania and hydrogenated sample are very similar. This indicates that the material has not changed significantly during the plasma treatment. However, as shown in Fig. 2b, a slight shift is observed of the Eg(1) maximum of 143.3 cm−1 registered for pure TiO2NTs to 145.7 cm−1 for H–TiO2NTs. This shift can be attributed to the increased number of oxygen vacancies in H–TiO2.31 Furthermore, a closer view of the Raman spectra reveals that the Eg(1) peak of hydrogen plasma-treated titania is broadened compared with that of pure TiO2. The spectral broadening is suggested to arise from modification32 of the crystal lattice resulting from oxygen vacancy, as oxygen vacancy defects are known to have a role in absorption of O2 molecules or can act as electron scavengers leading to retardation of charge recombination. The intensity of the anatase signal Eg(1) of the H–TiO2NTs is also much higher than that of pure TiO2, suggesting that the crystal size of TiO2 may be increased by hydrogen plasma treatment.33,34 According to Samsudin et al.,34 however, Ti3+ and oxygen vacancies have larger roles in the improved electrical properties of hydrogenated titania than do surface disorder or change in crystal size created during the hydrogenation process.
C)–O bond vibrations in the thiophene ring. Two peaks at 1495 cm−1 and 1561 cm−1 could be interpreted as asymmetric CC modes in polymer chains.29 The peaks 1366 and 1267 cm−1 are characteristic of vibrations of single-bonded carbon atoms in the polymer chain and in the thiophene ring, respectively. The oxyethylene ring deformations can be identified at 988, 571, and 442 cm−1.30 The Raman spectra registered for the pure titania and hydrogenated sample are very similar. This indicates that the material has not changed significantly during the plasma treatment. However, as shown in Fig. 2b, a slight shift is observed of the Eg(1) maximum of 143.3 cm−1 registered for pure TiO2NTs to 145.7 cm−1 for H–TiO2NTs. This shift can be attributed to the increased number of oxygen vacancies in H–TiO2.31 Furthermore, a closer view of the Raman spectra reveals that the Eg(1) peak of hydrogen plasma-treated titania is broadened compared with that of pure TiO2. The spectral broadening is suggested to arise from modification32 of the crystal lattice resulting from oxygen vacancy, as oxygen vacancy defects are known to have a role in absorption of O2 molecules or can act as electron scavengers leading to retardation of charge recombination. The intensity of the anatase signal Eg(1) of the H–TiO2NTs is also much higher than that of pure TiO2, suggesting that the crystal size of TiO2 may be increased by hydrogen plasma treatment.33,34 According to Samsudin et al.,34 however, Ti3+ and oxygen vacancies have larger roles in the improved electrical properties of hydrogenated titania than do surface disorder or change in crystal size created during the hydrogenation process.
 | ||
| Fig. 2 (a) Raman spectra of pristine and hydrogenated titania, pEDOT:PSS film, and composite material, (b) closer view of the Eg(1) peak for pristine and hydrogenated TiO2. | ||
UV-Vis spectroscopy was also used to characterize the optical properties of the obtained materials, and to determine the energy bandgap for H–TiO2 and TiO2 nanotubes. Fig. 3a and b shows the absorbance curves and the plot of Kubelka–Munk function vs. photon energy. Typical absorption in the UV region,35 the broad band in the range of 400–600 nm, was registered for both pure and H–TiO2 NTs, but the absorbance ability of hydrogenated titania is much higher compared with that of pure TiO2. The values of Ebg determined on the basis of the Tauc plot are 3.08 and 2.98 eV for pure and H–TiO2 NTs, respectively. The narrowing of bandgap energy was discussed in detail by Wang et al.:36 upon hydrogenation, the titania disorder shell loses lattice periodicity and breaks the octahedral symmetry of TiO6, with formation of a tail of the conduction band (CB), which leads to bandgap narrowing. Surface defects and reconstruction in titania nanocrystals lead to strong band tailing near the CB edge, whereas hydrogen atoms introduce localized states below the CB minimum. To summarize, enhanced absorbance in a visible region results from electronic transitions from the tailed conduction band, localized states of oxygen vacancies, and from mid-gap electronic states to the conducting band.
 | ||
| Fig. 3 (a) Absorbance spectra for titania nanotubes and composite material, (b) the Tauc plot for pure and hydrogenated TiO2NTs. | ||
For the pEDOT:PSS modified sample, a broad and intensive band between 500 and 700 nm is observed, resulting from the presence of polymer matrix.37 The increase in the absorbance ability of heterojunction in the visible range results from occurrence of π–π* transitions38 in the oxidized polymer matrix. The run of the absorbance curve registered for H–TiO2NTs/pEDOT:PSS indicates that the composite material should exhibit better photoactivity under visible light than its pure counterparts, therefore, it is especially useful for application in photovoltaic cells. A change in absorbance properties exhibited after polymer deposition is also clearly visible in the deep blue colour of the composite material (see Fig. S3†).
To investigate the change in the chemical state of elements on hydrogen plasma treatment, X-ray photoelectron spectroscopy measurements were performed. The XPS spectra recorded for titanium, oxygen, and carbon regions of hydrogenated titania are shown in Fig. 4, and Table 1 gives the position of each spectrum maximum. The respective spectra found for pure titania were described in our previous report.39 In contrast to unmodified TiO2, the region attributed to titania for H–TiO2 is characterized as two doublets containing both Ti2p1/2 and Ti2p3/2 signals. The highest maximum located at 459.07 eV is attributed to Ti(IV) in anatase crystalline phase.40 However, this value is shifted by 0.29 eV when compared with the respective peak recorded for pure titania. The small peaks at 457.20 eV for Ti2p3/2 and 462.62 eV for Ti2p1/2 result from the existence of Ti(III).41 The relation between the atomic content of Ti(IV) and Ti(III) is 14.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Appearance of the Ti(III) state is typical for samples treated in hydrogen plasma and suggests that oxygen vacancies are formed to maintain electrostatic balance.20
:1. Appearance of the Ti(III) state is typical for samples treated in hydrogen plasma and suggests that oxygen vacancies are formed to maintain electrostatic balance.20
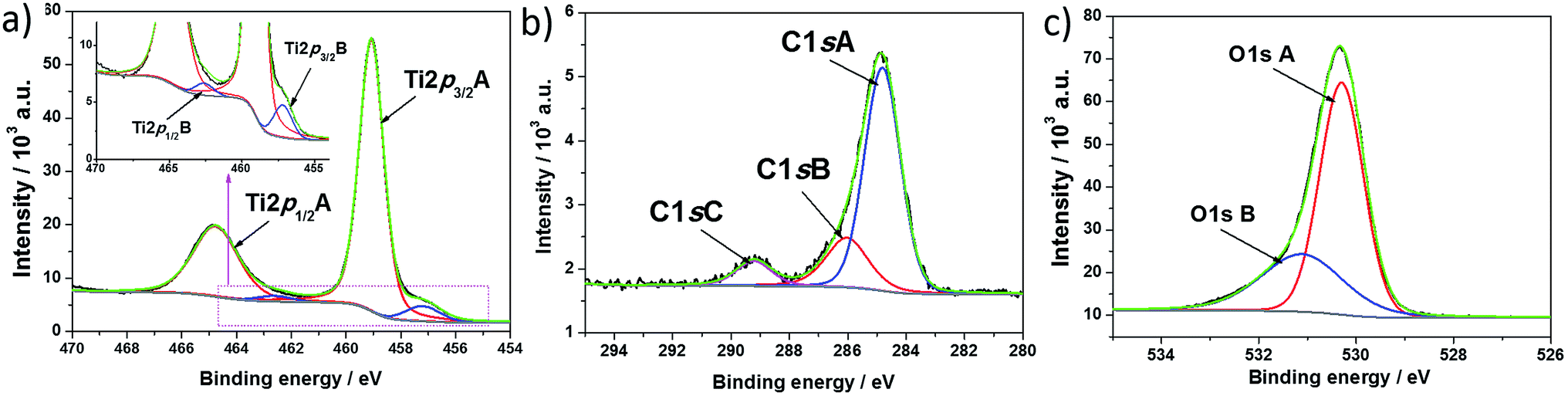 | ||
| Fig. 4 High resolution XPS spectra recorded at (a) titanium, (b) carbon, and (c) oxygen for H-TiO2 nanotubes. | ||
| Orbital | Symbol | Ebin/eV | at% |
|---|---|---|---|
| C1s | A | 284.81 | 10.31 |
| B | 286.33 | 3.00 | |
| C | 289.22 | 1.29 | |
| Ti2p | Ti2p3/2 A | 459.07 | 23.62 |
| Ti2p1/2 A | 464.79 | — | |
| Ti2p3/2 B | 457.20 | 1.66 | |
| Ti2p1/2 B | 462.62 | — | |
| O1s | A | 530.30 | 40 |
| B | 531.11 | 20.05 |
Concerning the oxygen region, the registered O1s signal could be fit by two single peaks located at 530.30 and 531.11 eV. The highest results from lattice oxygen Ti–O42 and is located at lower binding energy when compared with the pure sample. This probably results from a change in the oxidation state of partial titania species linked with oxygen atoms. The second peak found at 531.11 eV could be attributed to oxygen vacancy – Ti(III) type surface states.43 According to Wang et al.,44 oxygen vacancies have a critical role in visible light absorption, and therefore the absorbance ability of H–TiO2 is improved in comparison with pure TiO2.
As was registered for pure TiO2, a signal assigned to carbon species was also found in the hydrogenated sample. The C1s spectra were deconvoluted into three singlets with maxima located at 284.81 eV, 286.03 eV, and 289.22 eV. The first peak is typical of C–C bonds. According to the literature, the presence of such carbon species results from surface contamination originating from an anodization process performed in an organic electrolyte1 or from the XPS instrument itself.45 The second and third signal could be assigned to carbon linked to oxygen by single46 and double bonds,47 respectively.
As mentioned, the series of XPS measurements were performed with simultaneous etching of composite layers (see Fig. S4†). At the surface area, where polymer covers TiO2NT, signals attributed to oxygen and sulfur atoms reach their maximum values. After subsequent etching of H–TiO2NTs/pEDOT:PSS, the atomic contents of S and O decrease with simultaneous increase of signal attributed to titania species. However, from the 3 h up to the end of the etching procedure, the atomic contents of S, Ti, and O do not change significantly, which clearly reveals that the pEDOT:PSS matrix fills in the interior of the H–TiO2NTs layer and a three dimensional inorganic–organic interface is formed.
Electropolymerization analysis
As mentioned in the experimental section, deposition of conducting polymer was realized via potentiostatic electrochemical polymerization. The chronoamperometry curves for electropolymerization of pEDOT:PSS over a pure and hydrogenated titania layer are shown in Fig. 5. A huge difference was observed in the current density value and its shape. In the case of hydrogenated titania, an increase in the current is registered at the initial stage. Within about 15 s a current plateau is achieved, typical of already overlapped nuclei of a new phase. The whole polymerization process is realized within only 60 s, which may be a result of a short nucleation time.48,49 In the case of pure titania, an exponential current decay is observed. The shape of the current, which changes with time, is typical of early stages of polymerization before nuclei are overlapped.50 At a potential of 1.6 V vs. Ag/AgCl/0.1 M KCl, the electropolymerization occurs inefficiently on pristine titania and an extended time range of pEDOT:PSS deposition is needed for polymer film formation.51 Despite electropolymerization on pure TiO2 being carried out for an hour, uniform polymer deposition was not achieved, meaning comparison of electrochemical and photoelectrochemical properties between H–TiO2/pEDOT:PSS and TiO2/pEDOT:PSS composites was not possible. | ||
| Fig. 5 Current time transients for pEDOT:PSS electropolymerization at pure and hydrogenated TiO2NTs registered at E = 1.6 V vs. Ag/AgCl/0.1 M KCl (electrolyte: aqueous solution of 0.1 M NaPSS and 0.001 M EDOT). | ||
In contrast, electrode oxidation on H–TiO2 is fully realized by charge propagation and leads to formation of a conducting polymer film. Thus, it is suggested that the hydrogenation process activates the titania surface and facilitates monomer absorption and nucleation, with the entire electropolymerization occurring efficiently. However, further studies are required to prove changes in adsorption conditions on plasma treatment. At this stage of our investigation it can be postulated that facilitation results from a significant impedance decrease resulting from hydrogenation.
Electrochemical characterization of hydrogenated titania
The cyclic voltammetry curves registered for pure and hydrogenated titania are given in Fig. 6. In the investigated potential range, both samples deliver obvious pseudocapacitive characteristics that could be attributed to oxidation/reduction of surface hydroxyl groups.20 The observed low electrochemical activity of unmodified titania could be a result of its high resistance.52 The reduction peaks at around 0 V vs. Ag/AgCl/0.1 M KCl can be ascribed as an activity of intraband gap localized states.53 In contrast with pure TiO2NTs on hydrogen plasma treatment, the charging current density increases significantly, which is related to the capacitance enhancement and conductivity improvement.20,54 | ||
| Fig. 6 Cyclic voltammetry curves recorded for pristine and hydrogenated titania immersed in 0.5 M K2SO4, v = 50 mV s−1. | ||
To investigate sample resistance, electrochemical impedance measurements were conducted on pristine and hydrogenated titania materials. Spectra were collected at the rest potential as well as in the wide potential range. Fig. 7 shows that the spectra exhibit at least two time constants attributed to the response of the space charge layer in series with the Helmholtz layer at the electrolyte side. This concept state was proposed as a base for an analysis of obtained spectra and an electric equivalent circuit (EEQC), given in Fig. S5† with the value of each parameter listed in Table S1.† The same EEQC has been previously applied for pure and non-metal doped titania,27 and is similar to that proposed by other researchers.55,56 The goodness of the performed fitting procedure was in the order of 10−5 (the meaning of each element is given in a previous report27 and also in ESI†).
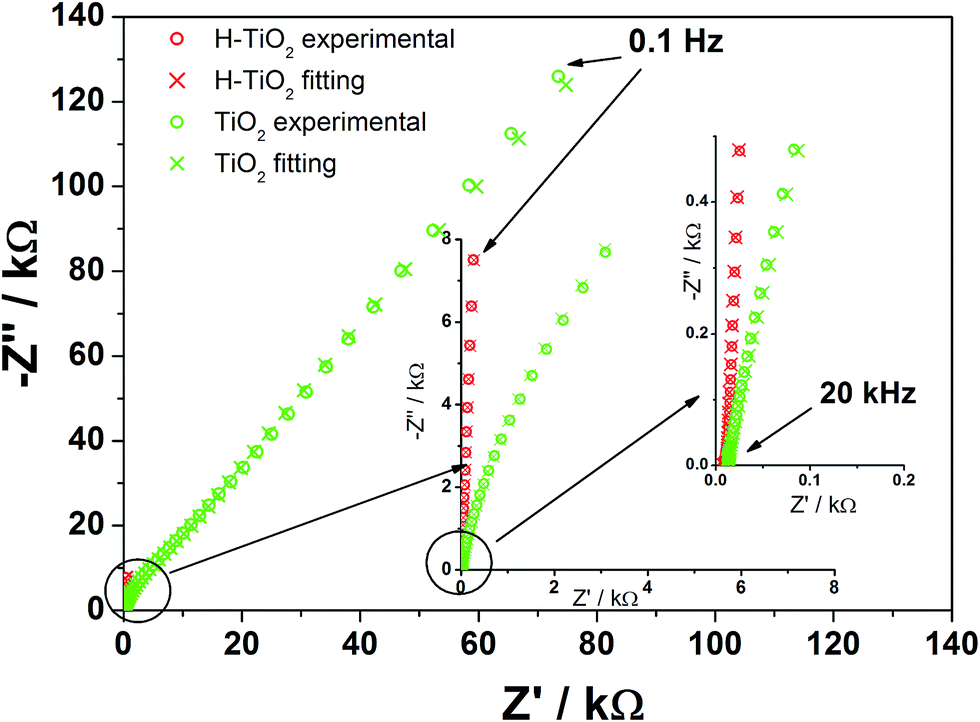 | ||
| Fig. 7 Impedance spectra registered for TiO2 and H–TiO2 nanotubes at the open circuit potential: EOCP (TiO2NTs) = +0.33 V, EOCP (H–TiO2NTs) = −0.09 V vs. Ag/AgCl/0.1 M KCl (electrolyte: 0.5 M K2SO4). | ||
When comparing the spectra registered at the open circuit potential (EOCP), an enormous decrease in R1 resistance of H–TiO2 in comparison with an untreated sample is observed, which suggests that charge transfer processes between tubular film and titanium support will be facilitated. The value of the CPE1 element does not change much on hydrogenation, whereas the value of the CPE2 element increases by two orders of magnitude. The inconsiderable increase of capacitance attributed to the bottom, planar oxide film is related to its low real surface area and electrochemical activity. On the other hand, enlargement of the tubular oxide capacity denoted for H–TiO2 could be explained in terms of higher donor concentration and anodic shift of the flatband potential when compared with pure TiO253 (as shown later).
Analysis of the impedance spectra recorded in the wide potential range allows for further analysis according to the Mott–Schottky relation, given as:
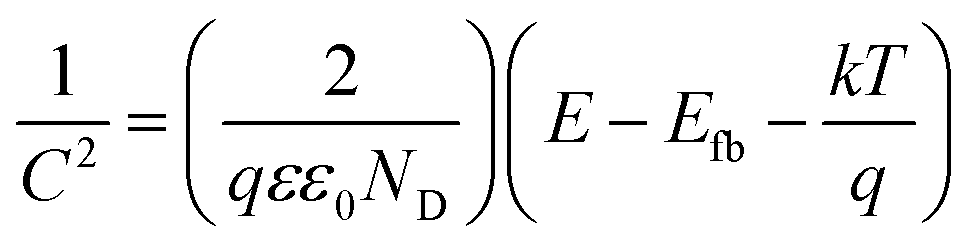 | (1) |
 | (2) |
Electrochemical properties of the composite material
Electrochemical studies were also conducted on the composite material, with cyclic voltammetry and electrochemical impedance spectroscopy measurements performed. CV curves and registered impedance spectra at the open circuit potential are shown in Fig. 8 and 9, respectively. The working electrode was polarized from rest potential in the anodic direction up to 0.7 V and back up to −0.7 V vs. Ag/AgCl/0.1 M KCl. Among tested materials, only the composite electrode exhibits symmetrical cathodic/anodic current peaks at +0.15 V vs. Ag/AgCL/0.1 M KCl and significantly higher current density when compared with that recorded for H–TiO2 nanotube or pure polymer film, illustrating improved electrochemical activity. The electrochemical impedance measurements performed on the composite material show its hybrid nature between pure polymeric and titania material. The shape of registered spectra for H–TiO2NT/pEDOT:PSS is similar to that of pEDOT:PSS alone,65 but the resistance is higher because of the presence of titania network. The EEQC proposed for titania samples was modified by the introduction of an additional arrangement of elements attributed to the polymer part (see Fig. S7†). The goodness of the proposed fitting equals 6.5 × 10−5, and the values for each element of EEQC are listed in Table S2.† When conducting polymer is present, the electronic conductivity of the whole composite results not only from the titania network, but also from charge propagation along the oxidized pEDOT chains. The action accruing at the oxidized chains could proceed according to the reactions:66| pEDOTx+ + xe− + PSS− → pEDOT + x(PSS−)sol |
| pEDOTx+(PSS−)x + xe− + C+ → pEDOT(PSS−)x(C+)x |
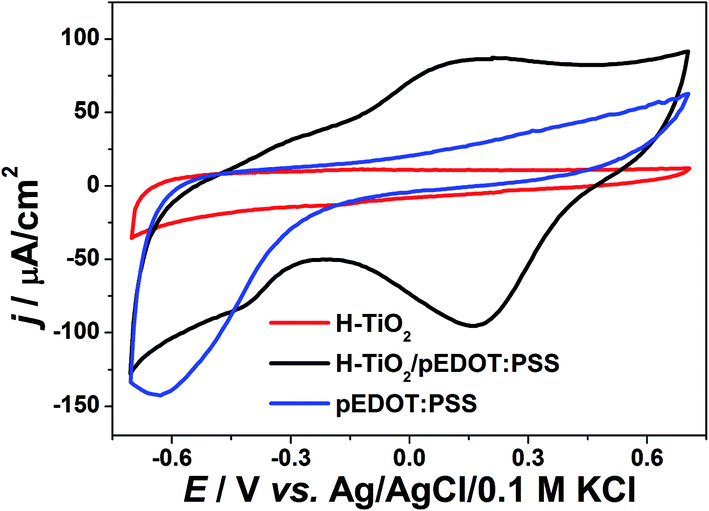 | ||
| Fig. 8 Cyclic voltammetry curves of composite material H–TiO2NTs/pEDOT:PSS and its pure counterparts (electrolyte: 0.5 M K2SO4, v = 50 mV s−1). | ||
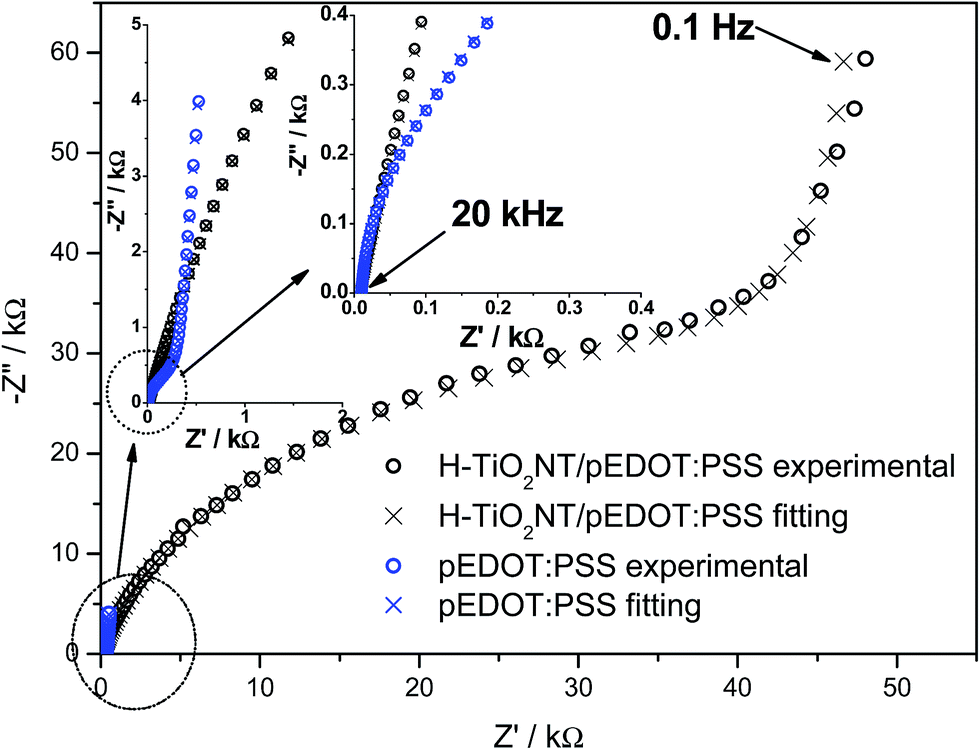 | ||
| Fig. 9 Impedance spectra registered for pEDOT:PSS and composite material: H–TiO2NTs/pEDOT:PSS at the open circuit potential: EOCP (pEDOT:PSS) = +0.27 V, EOCP (H–TiO2NTs/pEDOT:PSS) = +0.24 V vs. Ag/AgCl/0.1 M KCl (electrolyte: 0.5 M K2SO4). | ||
Interpretation of the majority of elements was given in the section concerning impedance of H–TiO2NTs. Here, R3 is attributed to resistance at the TiO2 nanotube/polymer interface and CPE3 reflects the pseudocapacitance of pEDOT:PSS.67 Because of the capacitive character exhibited by infiltrated polymer, the overall capacitance of the composite increases, which is in agreement with the shape of cyclic voltammetry curves. In summary, the registered impedance spectra show the double nature of H–TiO2NTs/pEDOT:PSS material and the impact of polymer is evident on improvement of composite electric properties. The decrease in overall resistance could be important in transfer of photogenerated charge carriers.
Photoelectrochemical activity
Photoelectrochemical properties of electrodes were investigated using the chronoamperometry technique recorded at constant potential under “chopped” simulated solar light illumination. The materials obtained were anodically polarized (E = 0.5 V vs. Ag/AgCl/0.1 M KCl) and immersed in 0.5 M K2SO4 aqueous electrolyte without any additives acting as a hole capturer. The chronoamperometry curves of H–TiO2NTs/pEDOT:PSS, and as reference: H–TiO2NTs, TiO2NTs, and pEDOT:PSS layer, are presented in Fig. 10, and the values of photocurrent density registered after 8 min of measurements are listed in Table 2.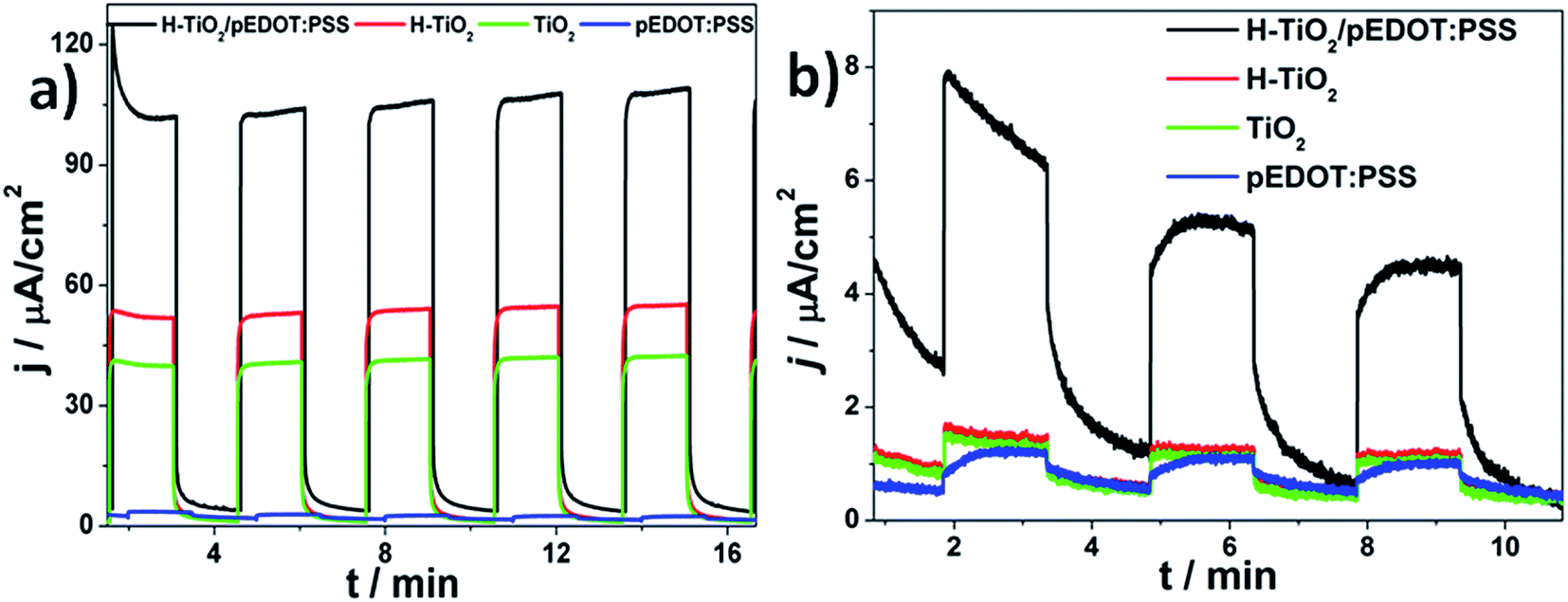 | ||
| Fig. 10 Transient photocurrent response of pure and hydrogenated titania, pure polymer, and H–TiO2NT/pEDOT:PSS composite material registered under (a) UV-Vis and (b) vis illumination at 0.5 V vs. Ag/AgCl/0.1 M KCl (electrolyte: 0.5 M K2SO4). | ||
| Electrode material | j (UV-Vis)/μA cm−2 | j (Vis)/μA cm−2 |
|---|---|---|
| H–TiO2NTs/pEDOT:PSS | 106 | 4.9 |
| H–TiO2 NTs | 54 | 0.9 |
| TiO2 NTs | 42 | 0.8 |
| pEDOT:PSS | 2 | 0.7 |
Among all tested materials, conducting polymer alone exhibits the lowest photoactivity that is typical of neat PEDOT.8,26 For H–TiO2 and TiO2 nanotubes, after 8 min of measurement duration, the photocurrent densities on UV-Vis irradiation reached 54 and 42 μA cm−2, respectively. Thus, a hydrogenated sample is characterized with almost 30% enhancement that results from a higher absorbance ability and improved electric properties when compared with pristine titania. It should also be noted that, despite hydrogenation treatment, titania response on illumination is stable and no current diminution is observed. After deposition of polymer, the obtained composite material is characterized by remarkable photoactivity, reaching ca. 106 μA cm−2 under UV-Vis illumination and 4.9 μA cm−2 when the electrode is illuminated by vis radiation. The observed enhancement is significantly higher under UV-Vis and Vis irradiation than that described for the heterojunction composed of pEDOT:PSS and titanium dioxide nanoparticles.68 Improved photoactivity of H–TiO2NTs/pEDOT:PSS in comparison with its pure counterparts results from formation of a p–n heterojunction when appropriate alignment of conduction and valence band positions of both p-type and n-type semiconductors allows for charge separation at the organic/inorganic heterojunction.69 According to Haring et al.,70 in the case of conducting polymer–TiO2 composite, there are three criteria for efficient hybrid bulk heterojunction: (a) Fermi level of TiO2 to be close to its conduction band, (b) the difference between conduction band and LUMO should equal at least 0.3 eV,71 and (c) Fermi level should be higher when compared with energy polarons level in the separated materials.72
On infiltration of titania by pEDOT:PSS, charge transfer from TiO2 would create positive polarons in the polymer matrix, resulting in formation of an electric field in the polymer that would assist photoelectron injection. Furthermore, in the presence of oxygen vacancies, the excess of negative charge on Ti(III) sites leads to formation of occupied deep traps below the conduction band.73 As demonstrated, unlike pristine titania, the proposed hydrogenation treatment of TiO2NTs results in partial reduction of Ti(IV) to Ti(III) and a decrease of the Fermi level of the flatband electrode, which could allow for spontaneous electron transfer from pEDOT:PSS to H–TiO2NTs. It should be noted that it is not just the favourable conditions for formation of electron polarons in H–TiO2NTs/pEDOT:PSS that are responsible for photocurrent increase of in the p–n junction. Other factors are also important in enhancement of photoconversion efficiency, such as improved absorbance ability in the visible range when compared with titania NTs or the pEDOT:PSS layer, and ordered morphology of the titania layer facilitating electron percolation and providing a large contact area for uniform distribution of polymer on the surface as well as along the tubes.74
Additionally, stability of H–TiO2NTs/pEDOT:PSS on prolonged illumination was also verified (see Fig. S8†). In contrast with our previous results on the TiO2NTs/pEDOT:PSS heterojunction14 or other titania-polymer heterojunctions,75,76 even over 50 min of illumination, no drop was observed in photoactivity. It should also be noted that on long-term irradiation the current density value slightly increases, because of oxidation of the polymer layer induced by photon absorption. The change in oxidation has an impact on the position of energy levels,77 and thus charge transfer at the organic/inorganic interface could proceed more efficiently. Electrochemical stability of the composite material was confirmed before and after illumination using cyclic voltammetry measurements. As shown in Fig. S6b,† the shape of the CV curve is unchanged, confirming the excellent photoresistivity of the organic–inorganic heterojunction.
Conclusions
In this study, the conducting polymer pEDOT:PSS was used as an organic sensitizer to form three-dimensional heterojunctions with ordered titania nanotubes. Infiltration of titania with the polymer was realized via electropolymerization carried out on titania electrode followed by treatment in hydrogen plasma. Spectroscopic and electrochemical measurements show that the proposed plasma processing leads to formation of Ti(III) species, enhancement of absorbance ability in the visible range, and conductivity and capacitance improvement when compared with pristine TiO2. On the basis of the impedance spectra analysis, flatband potential and donor density for TiO2 and H–TiO2NTs were estimated to be Efb = −0.16 V vs. Ag/AgCl/0.1 M KCl, ND = 6.45 × 1020 cm−3, and Efb = +0.11 V vs. Ag/AgCl/0.1 M KCl and ND = 2.27 × 1023 cm−3, respectively. Detailed elemental analysis has shown that hydrogen plasma facilitates uniform deposition of polymer matrix resulting in high surface area of the organic–metal oxide interface. The obtained composite material exhibits much higher capacitive current when compared with its pure counterparts, and the registered impedance spectra reflect its duplex polymer–metal oxide nature. Appropriate alignment of energy levels of p- and n-elements of the H–TiO2NTs/pEDOT:PSS heterojunction results in photocurrent density of 106 μA cm−2 under UV-vis and 4.9 under vis irradiation, approximately 2 and 5.6 times higher, respectively, than that registered for H–TiO2. To summarize, an H–TiO2NTs/pEDOT:PSS composite, offering the possibility of formation of a large p–n heterojunction, is as an attractive material for design of photovoltaic cells.Acknowledgements
This work received financial support from the Polish National Science Centre: Grant No. 2012/07/D/ST5/02269.References
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., 2011, 50, 2904 CrossRef CAS PubMed.
- D. Kowalski, D. Kim and P. Schmuki, Nano Today, 2013, 8, 235 CrossRef CAS.
- D. Wang, Y. Liu, B. Yu, F. Zhou and W. Liu, Chem. Mater., 2009, 21, 1198 CrossRef CAS.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3 CrossRef CAS.
- B. Chen, J. Hou and K. Lu, Langmuir, 2013, 29, 5911 CrossRef CAS PubMed.
- K. Siuzdak, M. Abbas, L. Vignau, M. Devynck, G. Dubacheva and A. Lisowska-Oleksiak, J. Appl. Phys., 2012, 112, 123110 CrossRef.
- Q. Chen and D. Xu, J. Phys. Chem. C, 2009, 113, 6310 CAS.
- J. Boucle, P. Ravirajan and J. Nelson, J. Mater. Chem., 2007, 17, 3141 RSC.
- Y. Jia, P. Xiao, H. He, J. Yao, F. Liu, Z. Wang and Y. Li, Appl. Surf. Sci., 2012, 258, 6627 CrossRef CAS.
- J. Luo, Y. Ma, H. Wang and J. Chen, Electrochim. Acta, 2015, 167, 119 CrossRef CAS.
- P. M. Dziewoński and M. Grzeszczuk, Electrochim. Acta, 2010, 55, 336 CrossRef.
- G. F. Samu, C. Visy, K. Rajeshwar, S. Sarker, V. R. Subramanian and C. Janáky, Electrochim. Acta, 2015, 151, 467 CrossRef CAS.
- S. H. Mujawar, S. B. Ambade, T. Battumur, R. B. Ambade and S. H. Lee, Electrochim. Acta, 2011, 56, 4462 CrossRef CAS.
- Y. Jia, P. Xiao, H. He, J. Yao, F. Liu, Z. Wang and Y. Li, Appl. Surf. Sci., 2012, 258, 6627 CrossRef CAS.
- K. Siuzdak, M. Sawczak and A. Lisowska-Oleksiak, Solid State Ionics, 2015, 271, 56 CrossRef CAS.
- H. Du, Y. Xie, C. Xia, W. Wang and F. Tian, New J. Chem., 2014, 38, 1284 RSC.
- C. Janaky, N. R. de Tacconi, W. Chanmanee and K. Rajeshwar, J. Phys. Chem. C, 2012, 116, 19145 CAS.
- D. Kowalski and P. Schmuki, Chem. Commun., 2010, 46, 8585 RSC.
- S. Xie, M. Gan, L. Ma, Z. Li, J. Yan, H. Yin, X. Shen, Z. Xu, J. Zhang and J. Hu, Electrochim. Acta, 2011, 56, 4462 CrossRef.
- C. Janaky and K. Rajeshwar, Prog. Polym. Sci., 2015, 43, 96 CrossRef CAS.
- J. Chen, Z. Xia, H. Li, Q. Li and Y. Zhang, Electrochim. Acta, 2014, 120, 408 CrossRef.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690 CrossRef CAS PubMed.
- H. Wu, C. Xu, J. Xu, L. Lu, Z. Fan, X. Chen, Y. Song and D. Li, Nanotechnology, 2013, 24, 455401 CrossRef PubMed.
- K. Siuzdak, M. Szkoda, M. Sawczak and A. Lisowska-Oleksiak, New J. Chem., 2015, 39, 2741 RSC.
- D. Kowalski and P. Schmuki, RSC Adv., 2013, 3, 2154 RSC.
- C. Janáky, G. Bencsik, A. Racz and C. Visy, Langmuir, 2010, 26, 13697 CrossRef PubMed.
- K. Siuzdak, M. Szkoda, M. Sawczak, A. Lisowska-Oleksiak, J. Karczewski and J. Ryl, RSC Adv., 2015, 5, 50379 RSC.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- A. Lisowska-Oleksiak, A. P. Nowak, M. Wilamowska, M. Sikora, W. Szczerba and C. Kapusta, Synth. Met., 2010, 160, 1234 CrossRef CAS.
- S. Garreau, G. Louarn and J. Buisson, Macromolecules, 1999, 32, 6807 CrossRef CAS.
- Z. Wang, C. Y. Yang, T. Q. Lin, H. Yin, P. Chen, D. Y. Wan, F. F. Xu, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Energy Environ. Sci., 2013, 6, 3007 CAS.
- X. B. Chen, L. Liu, P. Y. Yu, S. S. Mao, X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- A. Sasinka, T. Singh, S. Wang, S. AMthur and R. Kraehnert, J. Vac. Sci. Technol., A, 2015, 33, 01A152 Search PubMed.
- E. M. Samsudin, S. B. A. Hamid, J. C. Juan, W. J. Basirun and A. E. Kandjani, Appl. Surf. Sci., 2015, 359, 883 CrossRef CAS.
- Y. Mizukoshi, N. Ohtsu and N. Masahashi, Appl. Surf. Sci., 2013, 283, 1018 CrossRef CAS.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan and M. Jiang, Adv. Funct. Mater., 2013, 23, 5444 CrossRef CAS.
- C. Janáky, G. Bencsik, A. Rácz, C. Visy, N. R. de Tacconi, W. Chanmanee and K. Rajeshwar, Langmuir, 2010, 26, 13697 CrossRef PubMed.
- A. P. Nowak, M. Wilamowska and A. Lisowska-Oleksiak, J. Solid State Electrochem., 2010, 14, 263 CrossRef CAS.
- K. Siuzdak, M. Szkoda, A. Lisowska-Oleksiak, K. Grochowska, J. Karczewski and J. Ryl, Appl. Surf. Sci., 2015, 357, 942 CrossRef CAS.
- Q. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195 CrossRef CAS.
- X. Jiang, Y. Zhang, J. Jiang, Y. Rong, Y. Wang, Y. Wu and C. Pan, J. Phys. Chem. C, 2012, 116, 22619 CAS.
- X. Zhou, B. Jin, S. Zhang, H. Wang, H. Yu and F. Peng, Electrochem. Commun., 2012, 19, 127 CrossRef CAS.
- X. Wu, C. Xu, J. Xu, L. Lu, Z. Fan, X. Chen, Y. Song and D. Li, Nanotechnology, 2013, 24, 455401 CrossRef PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026 CrossRef CAS PubMed.
- Y. Li, G. Ma, S. Peng, G. Lu and S. Li, Appl. Surf. Sci., 2008, 254, 6831 CrossRef CAS.
- J. Yang, H. Bai, Q. Jiang and J. Lian, Thin Solid Films, 2008, 516, 1736 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and M. N. D. Brow, Carbon, 2005, 43, 153 CrossRef CAS.
- J. Xia, N. Masaki, K. Jiang and S. Yanagida, J. Mater. Chem., 2007, 17, 2845 RSC.
- A. A. Alamrio and R. L. Vieira, J. Chil. Chem. Soc., 2006, 51, 971 Search PubMed.
- M. E. G. Lyons, Transport and kinetics in electroactive polymers, Advances in Chemical Physics: Polymeric systems, ed. I. Progogine and S. A. Rice, Wiley & sons, 1996, vol. XCIV, p. 279 Search PubMed.
- M. Graczyk-Zajac, S. Y. Vassiliev, M. A. Vorotynsev and G. A. Tsirlina, J. Solid State Electrochem., 2010, 14, 2039 CrossRef CAS.
- F. Fabregat-Santiago, J. Mora-Seró, G. Garcia-Belmonte and J. Bisquert, J. Phys. Chem. B, 2003, 107, 758 CrossRef CAS.
- G. Boschloo and D. Fitzmaurice, J. Phys. Chem. B, 1999, 103, 2228 CrossRef CAS.
- H. Wu, D. Li, X. Zhu, C. Yang, D. Liu, X. Chen, Y. Song and L. Lu, Electrochim. Acta, 2014, 116, 129 CrossRef CAS.
- A. G. Muñoz, Q. Chen and P. Schmuki, J. Solid State Electrochem., 2007, 11, 1077 CrossRef.
- Z. Zhang, Z. Zhou, S. Nie, H. Wang, H. Peng, G. Li and K. Chen, J. Power Sources, 2014, 267, 388 CrossRef CAS.
- D. B. Bonham and M. E. A. Orazem, J. Electrochem. Soc., 1992, 139, 127 CrossRef CAS.
- X. Wang and J. Zhao, IEEE Trans. Nanotechnol., 2014, 14, 113 CrossRef.
- K. Gelderman, L. Lee and S. W. Donne, J. Chem. Educ., 2007, 84, 685 CrossRef CAS.
- C. Yang, Z. Wang, T. Lin, H. Yin, X. Lü, D. Wan and M. Jiang, J. Am. Chem. Soc., 2013, 135, 17831 CrossRef CAS PubMed.
- H. Tang, K. Prasad, R. Sanjines, P. E. Schmid and F. Levy, J. Appl. Phys., 1994, 75, 2042 CrossRef CAS.
- B. Chen, J. A. Beach, D. Maurya, R. B. Moore and S. Priya, RSC Adv., 2014, 4, 29443 RSC.
- H. Cui, W. Zhao, C. Yang, H. Yin, T. Lin, Y. Shan, Y. Xie, H. Gu and F. Hang, J. Mater. Chem. A, 2014, 2, 8612 CAS.
- X. Lu, G. Wang, Z. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690 CrossRef CAS PubMed.
- A. Lisowska-Oleksiak and A. P. Nowak, Solid State Ionics, 2008, 179, 72 CrossRef CAS.
- A. Lisowska-Oleksiak and A. Kupniewska, Solid State Ionics, 2003, 157, 241 CrossRef CAS.
- H. Du, Y. Zie, C. Xia, W. Wang and F. Tian, New J. Chem., 2014, 38, 1284 RSC.
- J. Baek, Y. Kim and E. Kim, Mol. Cryst. Liq. Cryst., 2008, 483, 275 CrossRef CAS.
- A. Arena, N. Donato, G. Saitta, G. Rizzo, G. Neri and G. Pioggia, J. Sol-Gel Sci. Technol., 2007, 43, 41 CrossRef CAS.
- A. J. Haring, S. R. Ahrenholtz and A. J. Morris, ACS Appl. Mater. Interfaces, 2014, 6, 4394 CAS.
- M. C. Scharber, D. Wuhlbacher, M. Koppe, P. Denk, C. W. Ardauf, A. J. Heeger and C. L. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- S. Braun, W. R. Salaneck and M. Fahlman, Adv. Mater., 2009, 21, 1450 CrossRef CAS.
- C. D. Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543 Search PubMed.
- L. Bertoluzzi, L. Badia-Bou, F. Fabregat-Santiago, S. Gimenez and J. Bisquert, J. Phys. Chem. Lett., 2013, 4, 1334 CrossRef CAS PubMed.
- Y. Jia, P. Xiao, H. He, J. Yao, F. Liu, Z. Wang and Y. Li, Appl. Surf. Sci., 2012, 258, 6627 CrossRef CAS.
- X. Zhang, F. Peng, H. Wang, H. Yu and Y. Fang, Chem. Commun., 2011, 47, 10323 RSC.
- Y. Xinag, M. Qian, D. Guo and G. Zhang, Chin. Phys. B, 2014, 23, 038504 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01986b |
| This journal is © The Royal Society of Chemistry 2016 |