
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detailed characterization of alterations in the lipid profiles during autophagic cell death of leukemia cells†
Jae Won
Lee
ab,
Haruka
Shinohara
c,
Jae Hun
Jung
a,
Hyuck Jun
Mok
a,
Yukihiro
Akao
c and
Kwang Pyo
Kim
*ad
aDepartment of Applied Chemistry, College of Applied Science, Kyung Hee University, Yongin, 446-701, Republic of Korea. E-mail: kimkp@khu.ac.kr
bDepartment of Herbal Crop Research, National Institute of Horticultural and Herbal Science, RDA, Eumseong 369-873, Republic of Korea
cUnited Graduate School of Drug Discovery and Medical Information Sciences, Gifu University, 1-1 Yanagido, Gifu 501-1193, Japan
dThe Institute of Natural Science, College of Applied Science, Kyung Hee University, Yongin, 446-701, Republic of Kore
First published on 14th March 2016
Abstract
In this study, a lipidomics approach based on UPLC-QqQ/MS was applied to profile various lipids in human leukemia cells undergoing autophagic cell death (ACD). Our previous study indicated that AIC-47, a 3-decenoic acid derivative, induced ACD in cancer cells that was associated with lipophagy. To understand the altered metabolism of lipids during ACD, 23 lipid classes were profiled in AIC-47 – treated cancer cells. Using the optimal UPLC conditions, individual lipid species were well separated in 30 min. By multiple reaction monitoring the 397 individual lipid species were successfully identified and quantified. 14 classes of lipid—TG, DG, PS, PG, PI, PA, LPC, LPE, LPS, LPG, LPI, Cer, Sa, and Cer1P—were upregulated and 3 classes—ChE, PC, and LPA—were downregulated in the ACD-induced cells compared to the control (P ≤ 0.05 by t-test). Other classes, such as PE, SM, dCer, So, So1P, and Sa1P, showed no changes. These results indicate that lipid metabolism of ACD is related to the mechanism of autophagy.
Introduction
For effective treatment of cancer it is critical to investigate specific molecules with anticancer effects.1,2 10-Hydroxy-2-decenoic acid, which is a component of royal jelly, shows anticancer activity.3 In our previous study, various decenoic acid derivatives were synthesized and screened.4 As a result, several 3-decenoic acid derivatives were found to show potent anticancer activity; in particular, AIC-47 induced autophagic cell death (ACD) in human leukemia K562 cells. As several anticancer drugs have been reported to induce ACD in cancer cells5,6 it is important to understand the mechanism of ACD.Cancer cells have often been found to have lower autophagic capacity than their normal counterparts.7 Autophagy in cancer cells is activated in response to various cellular stresses such as nutrient and growth factor starvation,8 and by therapeutic treatment.9 In such circumstances the dying cells display a large-scale accumulation of autophagic vacuoles, and this phenomenon led to the classification of ACD, which is a form of programmed cell death.10 However, this definition is under debate in different scientific communities because autophagy is generally thought of as a survival mechanism.11
Morphologically, the autophagosomes in cancer cells treated with AIC-47 contain lipid droplets (LDs).4 Several studies have characterized the roles of LDs as cellular stores of neutral lipids as well as a lipid source to support autophagic membrane formation.12,13 LDs are also associated with a specific type of autophagy called lipophagy that regulates lipid metabolism.14,15 In particular, lipophagy has the potential to regulate cellular energy homeostasis as well as lipid content.16
In this study, we performed lipid profiling of ACD-induced cancer cells to investigate the correlation between lipids and ACD. Lipid profiling is a critical method to study lipid functions in various biological samples.17,18 The roles of several lipids in the control of autophagy have been previously reported;19 however, there are still many other lipids that have not yet been characterized in autophagic cells. Furthermore, this study is the first evaluation of lipid alterations in cancer cells in the state of ACD induced by a decenoic acid derivative.
Experimental
Reagents
HPLC-grade methanol, acetonitrile, water, and 2-propanol were purchased from J. T. Baker (Avantor Performance Material, Inc, PA, USA). HPLC-grade formic acid was purchased from Fluka Analytical, Sigma Aldrich Chemie GmbH, Steinheim, Germany. Chloroform and ammonium formate were purchased from Sigma-Aldrich, St. Louis, MO, USA. Lipid standards used in this study were as follows: PC (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–10:0), PC (12:0–12:0), PE (10:0–10:0), PS (10:0–10:0), PG (10:0–10:0), PI (8:0–8:0), PA (10:0–10:0), LPC (13:0), LPE (14:0), LPS (17:1), LPG (14:0), LPI (13:0), LPA (14:0), SM (d18:1–12:0), Cer (d18:1–12:0), dCer (d18:1–12:0) So (d17:1), Sa (d17:0), Cer1P (d18:1–12:0), So1P (d17:1), and Sa1P (d17:0), all purchased from Avanti Polar Lipids, Inc. TG (11:1–11:1–11:1), DG (8:0–8:0), and ChE (10:0) were purchased from Larodan Fine Chemicals AB (Malmö, Sweden).
:0–10:0), PC (12:0–12:0), PE (10:0–10:0), PS (10:0–10:0), PG (10:0–10:0), PI (8:0–8:0), PA (10:0–10:0), LPC (13:0), LPE (14:0), LPS (17:1), LPG (14:0), LPI (13:0), LPA (14:0), SM (d18:1–12:0), Cer (d18:1–12:0), dCer (d18:1–12:0) So (d17:1), Sa (d17:0), Cer1P (d18:1–12:0), So1P (d17:1), and Sa1P (d17:0), all purchased from Avanti Polar Lipids, Inc. TG (11:1–11:1–11:1), DG (8:0–8:0), and ChE (10:0) were purchased from Larodan Fine Chemicals AB (Malmö, Sweden).
Cell culture and treatment
The human leukemia cell line K562 was cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) containing 10% (v/v) FBS and 1% penicillin in controlled cell culture conditions of 5% CO2 and 37 °C. For treatment with AIC-47, 1 μL of AIC-47 was dissolved in 300 μL of dimethyl sulfoxide (DMSO), resulting in a 10 mM solution. This solution was dissolved in 1 mL culture medium to give a final concentration of 10 μM AIC-47. ACD was induced after 48–72 h. After treatment, the cell pellet was collected from each culture dish and the number of cells was counted using a hemocytometer. The total number of cells was 2.55 × 106 for AIC-47 treated and 4.45 × 106 for non-treated cells. Lipid extraction was performed using 1.275 × 106 cells from each group.Sample preparation
Each lipid standard was prepared by dissolution in methanol:chloroform (1:1). These standard solutions were stored at −20 °C and diluted to the desired concentration for use. For the lipid extraction of cell lines, two-step extractions including neutral and acidic extractions were performed. First, for neutral extraction, the cell pellet was added to 990 μL of chloroform/methanol (1:2, v/v) and 10 μL of 1 μg mL−1 of lipid standards including TG (11:1–11:1–11:1), DG (8:0–8:0), ChE (10:0), PC (10:0–10:0), PE (10:0–10:0), PS (10:0–10:0), PG (10:0–10:0), PI (8:0–8:0), PA (10:0–10:0), LPC (13:0), LPE (14:0), LPS (17:1), LPG (14:0), LPI (13:0), LPA (14:0), SM (d18:1–12:0), Cer (d18:1–12:0), dCer (d18:1–12:0), So (d17:1), Sa (d17:0), Cer1P (d18:1–12:0), So1P (d17:1), and Sa1P (d17:0) as IS. The sample was vortexed for 30 s every 3 min. After centrifugation (13800 × g, 2 min at 4 °C), 950 μL of supernatant was transferred to a new Eppendorf tube. Second, for acidic extraction the remaining pellet was resuspended in 750 μL chloroform/methanol/37% (1 N) HCl (40:80:1, v/v/v) and incubated for 15 min at room temperature with vortexing for 30 s every 5 min. The tube was transferred to ice, and 250 μL cold chloroform and 450 μL cold 0.1 M HCl was added followed by 1 min vortexing and centrifugation (6500 × g, 2 min at 4 °C). The lower organic phase was transferred to a new tube.20
TMSD methylation
A solution of trimethylsilyldiazomethane (TMSD) (2 mol L−1) in hexane was added to the lipid extracts from the cell lines and the lipid standard samples dissolved in methanol to obtain yellow-colored solutions. After vortexing for 30 s, methylation was performed at 37 °C for 15 min. Addition of glacial acetic acid (6 μL) quenched the methylation and yielded colorless samples. The samples were subjected to LC/MS.UPLC-QqQ/MS analysis
Quantitative lipid profiling was performed by 6490 Accurate-Mass Triple Quadrupole (QqQ) LC-MS coupled to a 1200 series HPLC system (Agilent Technologies, Wilmington, DE, USA) with a Hypersil GOLD column (2.1 × 100 mm ID; 1.9 μm, Thermo Science). This technique provides high sensitivity by iFunnel technology that consists of three components: Agilent Jet Stream technology, a hexabore capillary, and a dual ion funnel. The temperature of the column oven and sample tray was set to 40 °C and 4 °C, respectively. Solvent A consisted of an acetonitrile–methanol–water mixture (19:19:2) with 20 mmol L−1 ammonium formate and 0.1% (v/v) formic acid, and solvent B consisted of 2-propanol with 20 mmol L−1 ammonium formate and 0.1% (v/v) formic acid. The gradient elution program was as follows: 0–5 min, B 5%; 5–15 min, B 5–30%; 15–22 min, B 30–90%; 22–25 min, B 90%; 25–26 min, 90–5%; 26–30 min, B 5%. The flow rate was 250 μL min−1 and the injection volume was 2 μL for each run. Total run time was 30 min for each analysis. All acquisition methods used the following parameters: 3500 V positive mode of capillary voltage, 3000 V negative mode of capillary voltage, sheath gas flow of 11 L min−1 (UHP nitrogen) at 200 °C, drying gas flow of 15 L min−1 at 150 °C, nebulizer gas flow at 25 psi. Multiple reaction monitoring (MRM) conditions including transition and MS/MS collision energy were optimized to analyze target lipids in individual samples.
Result and discussion
Construction of a lipid profiling method based on UPLC/QqQ-MS
For the profiling of various lipids, we constructed a MRM method based on UPLC-QqQ/MS. First, 23 lipid standards were used to optimize the MRM conditions for TG, DG, ChE, PC, PE, PS, PG, PI, PA, LPC, LPE, LPS, LPG, LPI, LPA, SM, Cer, dCer, So, Sa, Cer1P, So1P, and Sa1P. In a previous study, TMSD methylation was applied for the sensitive profiling of several highly acidic lipids.21 Therefore, we used TMSD methylation for the analysis of PS, PI, PA, LPS, LPI, LPA, Cer1P, So1P, and Sa1P. By MS scan, the adducted ions of each lipid were confirmed to construct the MRM transition (precursor m/z [Q1] > product m/z [Q3]). As a result, in the positive ion mode of ESI,† each lipid was detected with two adducted ions, a hydrogen ion (H+; m/z 1.01) and an ammonium ion (NH4+; m/z 18.03). For example, TG, DG, ChE, and PG were detected as [M + NH4]+ ions. Furthermore, PC, PE, LPC, LPE, LPG, SM, Cer, dCer, So, Sa, and several methylated lipids such as PS, PI, PA, LPS, LPI, LPA, Cer1P, So1P, and Sa1P were detected as [M + H]+ ions. By the product ion scan, m/z of one product ion with high intensity was determined as the Q3 of transition, and the MS/MS collision energy was optimized (Table S1†).Next, validation of lipid analysis based on MRM was performed to estimate the performance of lipid quantification. Each lipid standard was analyzed six times with an internal standard (IS), PC (12:0–12:0), and the relative standard deviation (RSD) (%) was calculated. The RSDs (%) of the relative retention times (RTs) and relative peak areas were smaller than 1.5% and 8.9%, respectively. The method showed high reproducibility, and the correlation (R2) in each lipid analysis was at least 0.9803, indicating high reliability. The limits of detection (LODs) of each lipid standard were also listed (Table S2†).22
Lipid profiling of control and AIC-47 – treated leukemia cells
Next, we performed lipid profiling of control and AIC-47 – treated leukemia cells. The ability of AIC-47 to induce ACD has already been demonstrated in a previous study.4 To characterize in detail the lipid content of ACD-induced leukemia cells, we analyzed three neutral lipids (TG, DG, ChE), six phospholipids (PC, PE, PS, PG, PI, PA), six lysophospholipids (LPC, LPE, LPS, LPG, LPI, LPA), and eight sphingolipids (SM, Cer, dCer, So, Sa, Cer1P, So1P, Sa1P). Neutral lipids have functions in energy storage23,24 and phospholipids, lysophospholipids, and sphingolipids have roles in cellular membrane components and signaling transduction.25,26 Thus, detailed profiling of these lipids is useful to not only to estimate the lipid content but also to understand their functional roles in ACD. Using the optimal UPLC-QqQ/MS conditions, various lipids including 71 TGs, 19 DGs, 11 ChEs, 59 PCs, 36 PEs, 29 PSs, 21 PGs, 19 PIs, 33 PAs, 15 LPCs, 11 LPEs, 2 LPSs, 6 LPGs, 6 LPIs, 17 LPAs, 14 SMs, 13 Cers, 3 dCers, 2 So, Sa, 7 Cer1Ps, So1P, and Sa1P were successfully analyzed in AIC-47 – treated leukemia cells (Table S3†). The total ion chromatogram of lipids is represented in Fig. S1.† Phospholipids, lysophospholipids, sphingolipids, and DGs were eluted from 0 to 15 min, whereas TGs, ChEs, and methylated PIs were eluted from 15 to 21 min. Using an octadecylsilyl silica column lipid species were separated according to their fatty acid composition, including different carbon numbers and double bond numbers. Each IS was used to confirm the RT of lipids. Each lipid species was assigned according to its RT in the chromatogram.Detailed characterization of altered lipids in ACD-induced cancer cells
In the ACD-induced leukemia cells and controls, a total of 397 individual lipid species were analyzed by UPLC-QqQ/MS. First, we applied principal component analysis (PCA) to differentiate ACD-induced leukemia cells and controls. In the PCA score plot, the two groups were separated well with 77.2% total variances including principal component (PC) 1 (64.5%) and PC 2 (12.7%) (Fig. 1A). This indicated that ACD induced the alteration of various lipids in leukemia cells. In the volcano plot, 56 lipid species were selected as the differently regulated lipids with fold change >2 and p value < 0.05 (Fig. 1B, Table 1). We found that several lipids were up or down regulated by ACD.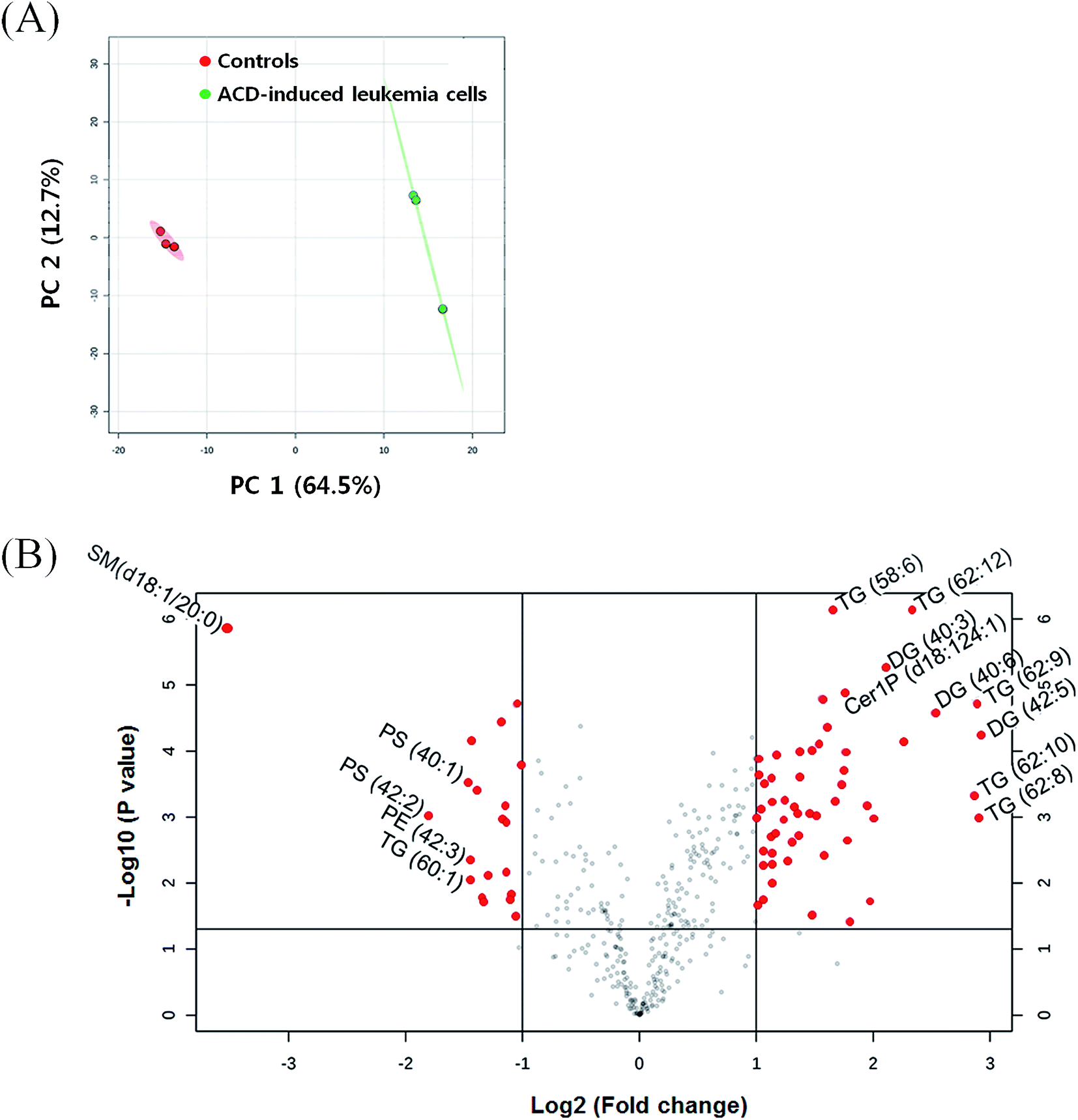 | ||
| Fig. 1 Principal component analysis score plot of controls (red) and ACD-induced leukemia cells (green) (A) and the volcano plot representing differently regulated lipids in ACD-induced leukemia cells compared to controls (fold change > 2, p value < 0.05) (B). | ||
| No. | Lipids | log2 (fold change) |
p value | No. | Lipids | log2 (fold change) |
p value |
|---|---|---|---|---|---|---|---|
| 1 | TG (56:5) |
1.37 | 0.0001 | 29 | PC (32:0) |
1.01 | 0.0001 |
| 2 | TG (56:6) |
1.60 | 0.00004 | 30 | PC (32:2) |
1.17 | 0.0001 |
| 3 | TG (58:4) |
1.14 | 0.002 | 31 | PC (34:3) |
1.00 | 0.001 |
| 4 | TG (58:5) |
1.78 | 0.002 | 32 | PC (36:7) |
−1.05 | 0.00002 |
| 5 | TG (58:6) |
1.65 | 0.000001 | 33 | PE (34:0) |
1.47 | 0.0001 |
| 6 | TG (60:1) |
−1.44 | 0.009 | 34 | PE (38:5) |
−1.18 | 0.00004 |
| 7 | TG (60:11) |
2.00 | 0.001 | 35 | PE (42:3) |
−1.44 | 0.004 |
| 8 | TG (60:12) |
1.48 | 0.03 | 36 | PE (42:9) |
1.51 | 0.0009 |
| 9 | TG (60:2) |
−1.16 | 0.001 | 37 | PS (34:0) |
1.76 | 0.0001 |
| 10 | TG (62:11) |
2.26 | 0.00007 | 38 | PS (34:2) |
1.14 | 0.01 |
| 11 | TG (62:12) |
2.34 | 0.000001 | 39 | PS (38:3) |
−1.11 | 0.016 |
| 12 | TG (62:6) |
1.02 | 0.02 | 40 | PS (38:5) |
−1.34 | 0.017 |
| 13 | TG (62:7) |
1.13 | 0.003 | 41 | PS (40:3) |
−1.34 | 0.018 |
| 14 | TG (62:8) |
2.90 | 0.001 | 42 | PS (40:6) |
−1.29 | 0.008 |
| 15 | TG (62:9) |
2.88 | 0.00002 | 43 | PS (42:2) |
−1.80 | 0.0009 |
| 16 | TG (64:11) |
1.58 | 0.004 | 44 | PS (42:5) |
−1.14 | 0.007 |
| 17 | TG (64:12) |
1.16 | 0.002 | 45 | PS (42:7) |
−1.10 | 0.015 |
| 18 | DG (36:0) |
1.06 | 0.003 | 46 | PG (34:3) |
1.27 | 0.0045 |
| 19 | DG (40:3) |
2.11 | 0.000006 | 47 | PG (34:3) |
1.27 | 0.0045 |
| 20 | DG (40:6) |
2.52 | 0.00003 | 48 | PI (28:0) |
1.30 | 0.0023 |
| 21 | DG (42:5) |
2.93 | 0.00006 | 49 | PA (26:3) |
1.53 | 0.00008 |
| 22 | SM (d18:1–18:0) |
1.05 | 0.02 | 50 | PA (28:2) |
−1.05 | 0.03 |
| 23 | SM (d18:1–20:0) |
−3.53 | 0.000001 | 51 | PA (34:0) |
1.23 | 0.001 |
| 24 | SM (d18:1–20:1) |
1.36 | 0.002 | 52 | PA (36:3) |
1.56 | 0.00001 |
| 25 | Cer1P (d18:1–22:0) |
1.97 | 0.02 | 53 | PA (40:6) |
−1.43 | 0.00007 |
| 26 | Cer1P (d18:1–24:0) |
1.13 | 0.004 | 54 | PA (42:9) |
−1.14 | 0.001 |
| 27 | Cer1P (d18:1–24:1) |
1.76 | 0.00001 | 55 | Cer (d18:1–20:1) |
1.79 | 0.038 |
| 28 | Sa (d18:0) |
1.13 | 0.005 | 56 | Cer (d18:1–22:1) |
1.06 | 0.005 |
Next, we performed the quantification of each lipid class to find the lipid alterations induced by ACD. From the MRM data, the peak area of each lipid was calculated and normalized to the IS peak area (lipid species peak area/IS peak area). The quantified values of each species were then summed to obtain the total amount of each lipid class. For example, in the case of TG, the normalized values of 71 individual TG species were summed to calculate the total amount of TG in the sample. Next, we applied the t-test for the comparison of ACD-induced leukemia cells and controls (significance at P ≤ 0.05). As a result, the quantitative alteration of 23 lipid classes was in the ACD-induced leukemia cells (Table 2). Compared to the control, 14 classes—TG, DG, PS, PG, PI, PA, LPC, LPE, LPS, LPG, LPI, Cer, Sa, and Cer1P—were upregulated, and 3 classes—ChE, PC, and LPA—were downregulated in the ACD-induced leukemia cells. Other classes, such as PE, SM, dCer, So, So1P, and Sa1P, showed no changes. These lipid alterations might be associated with the autophagy that triggers ACD. Therefore, we tried to match our lipid profiling data of ACD-induced cells with previous reports about the various roles of lipids in autophagy.
| No. | Lipids | p value | Fold change (AIC-47/control) | Quantitative alteration |
|---|---|---|---|---|
| 1 | TG | 0.015 | 1.38 | Up |
| 2 | DG | 0.012 | 1.83 | Up |
| 3 | ChE | 0.032 | 0.57 | Down |
| 4 | PC | 0.006 | 0.88 | Down |
| 5 | PE | 0.84 | 0.99 | — |
| 6 | PS | 0.002 | 1.74 | Up |
| 7 | PG | 0.007 | 1.42 | Up |
| 8 | PI | 0.017 | 1.57 | Up |
| 9 | PA | 0.0009 | 1.71 | Up |
| 10 | LPC | 0.0007 | 1.35 | Up |
| 11 | LPE | 0.0099 | 1.16 | Up |
| 12 | LPS | 0.0005 | 5.54 | Up |
| 13 | LPG | 0.0085 | 1.23 | Up |
| 14 | LPI | 0.041 | 1.50 | Up |
| 15 | LPA | 0.0003 | 0.55 | Down |
| 16 | SM | 0.72 | 1.08 | — |
| 17 | Cer | 0.038 | 1.21 | Up |
| 18 | dCer | 0.12 | 1.50 | — |
| 19 | So | 0.11 | 1.41 | — |
| 20 | Sa | 0.015 | 2.54 | Up |
| 21 | Cer1P | 0.00011 | 1.21 | Up |
| 22 | So1P | 0.28 | 0.86 | — |
| 23 | Sa1P | 0.33 | 1.17 | — |
In our previous study we found that AIC-47 – treated cancer cells showed LDs together with autophagosomes.4 Several lipids, including TG, DG, ChE, PC, and PE, were critical components of these LDs. In the treated cells, TG and DG were upregulated, and ChE was downregulated. It was previously reported that AIC-47 induces a switch from glycolysis to the TCA cycle through switching from pyruvate kinase M (PKM)2 to PKM1, suggesting that increased generation of citric acid promotes synthesis of fatty acid and TG.27 Furthermore, ChE is known to inhibit autophagy by activating p38 MAPK in macrophages.28 Thus, AIC-47 may inactivate p38 MAPK and induce downregulation of ChE, leading to autophagy. The LDs generated by AIC-47 may consist of mainly TG and DG.
PC and PE, which are the main components of the LD surface,29 were not upregulated. The downregulation of PC and no change of PE may be related to the PA/DG/protein kinase C (PKC) signaling cascade and elongation of isolation membranes, respectively. It was previously reported that PKC signaling has a critical role in generation of autophagy.30,31 In general, phospholipase D (PLD), which is a positive modulator of autophagy, hydrolyzes PC to PA. DG is produced from PA by PA phosphatase (PAP) and mediates the downstream stimulation of PKC, which can in turn activate autophagy by dissociating the Bcl-2 and Beclin 1 complex and by stimulating NADPH oxidase.32 Our results also showed downregulation of PC and upregulation of PA and DG in the AIC-47 – treated cells. This might indicate that lipid metabolism for PKC signaling is related to autophagy (Fig. 2A). During autophagy, PE has critical roles in the elongation and closure of the isolation membrane19 and an artificial increase in PE levels increases cellular autophagic flux.33 PE is generally produced by decarboxylation of PS. The upregulated PS in the AIC-47 – treated cells might be used to produce PE for the isolation membrane. Furthermore, PE might show no changes because it may be dissociated from the surface of autophagosome membranes (Fig. 2B).
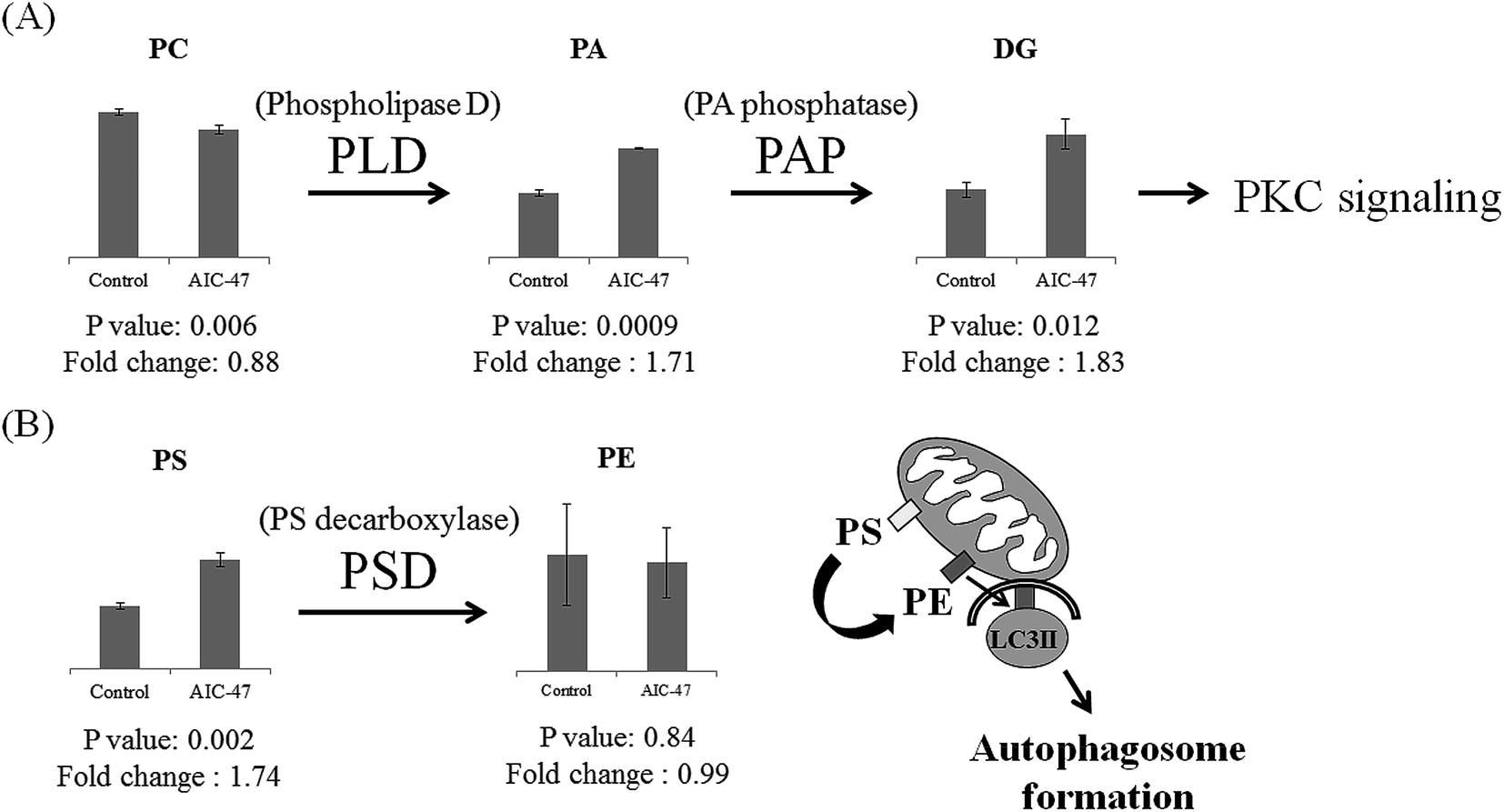 | ||
| Fig. 2 The lipid metabolism of autophagic cell death. (A) Quantitative alteration of PC, PA, and DG related to PKC signaling. (B) PE, synthesized from PS, has critical roles in autophagosome formation and closure. PA, phosphatidic acid; PC, phosphatidylcholine; DG, diacylglycerol; PKC, protein kinase C; PE, phosphatidylethanolamine; PS, phosphatidylserine. | ||
Upregulated PI is also related to PI3K class III signaling, which is essential for induction of autophagy. In our previous study we observed downregulation of Bcl-2 and upregulation of Beclin-1, indicating that PI3K class III signaling is activated by treatment with AIC-47.27 PI-3-phosphate (PI3P) generated from PI is also reported to be an important inducer of autophagy.19 In neutrophils, LPS activates reactive oxygen species (ROS)34 production through interaction with PKC. Upregulated LPS by AIC-47 may be associated with ROS generation and autophagy if the ROS production mechanism in leukemia cells is the same as that in neutrophils. Furthermore, downregulated LPA is known to inhibit autophagy.35 Previous studies have also reported that Cer can activate autophagy by inhibiting the phosphorylation of Akt, reducing the activation of mTOR, and upregulating Beclin 1 function.36,37
Conclusions
A lipidomics approach based on UPLC-QqQ/MS was used to characterize the altered metabolism of various lipids in ACD-induced human leukemia cells. Using the optimal UPLC and MRM, 397 individual species of 23 lipid classes were successfully analyzed. In the comparison of ACD-induced cells and controls, statistical data analysis indicated that ACD causes altered lipid metabolism in cancer cells. Upregulation or downregulation of lipids that have known roles in autophagy demonstrated that the lipid metabolism of ACD is correlated to autophagy. Furthermore, the induction of simultaneous alteration of other lipids by ACD is well described. Finally, the novel technique based on UPLC-QqQ/MS shows promising applications for comprehensive lipid profiling as well as for revealing and elucidating lipid alterations in a certain biological system such as ACD.Abbreviations
| ACD | Autophagic cell death |
| LDs | Lipid droplets |
| MRM | Multiple reaction monitoring |
| IS | Internal standard |
| TG | Triacylglycerol |
| DG | Diacylglycerol |
| ChE | Cholesterylester |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PS | Phosphatidylserine |
| PG | Phosphatidylglycerol |
| PI | Phosphatidylinositol |
| PA | Phosphatidic acid |
| LPC | Lysophosphatidylcholine |
| LPE | Lysophosphatidylethanolamine |
| LPS | Lysophosphatidylserine |
| LPG | Lysophosphatidylglycerol |
| LPI | Lysophosphatidylinositol |
| LPA | Lysophosphatidic acid |
| SM | Sphingomyelin |
| Cer | Ceramide |
| dCer | Dihydroceramide |
| So | Sphingosine |
| Sa | Sphinganine |
| Cer1P | Ceramide-1-phosphate |
| So1P | Sphingosine-1-phosphate |
| Sa1P | Sphinganine-1-phosphate |
| TMSD | Trimethylsilyldiazomethane |
| RSD | Relative standard deviation |
| LODs | Limits of detection |
| PCA | Principal component analysis |
| PKM | Pyruvate kinase M |
| PKC | Protein kinase C |
| PLD | Phospholipase D |
| PAP | Phosphatidic acid phosphatase |
| PI3P | Phosphatidylinositol-3-phosphate |
| ROS | Reactive oxygen species |
| mTOR | Mammalian target of rapamycin |
Acknowledgements
This work was supported by the Bio and Medical Technology Development program (project no. 2012M3A9B6055305) through the National Research Foundation of Korea funded by the Korean Ministry of Education, Science and Technology (to K. P. K.), Korea.Notes and references
- S. L. Holbeck, J. M. Collins and J. H. Doroshow, Mol. Cancer Ther., 2010, 9, 1451–1460 CrossRef CAS PubMed.
- V. Srivastava, A. S. Negi, J. Kumar, M. Gupta and S. P. Khanuja, Bioorg. Med. Chem., 2005, 13, 5892–5908 CrossRef CAS PubMed.
- G. F. Townsend, W. H. Brown, E. E. Felauer and B. Hazlett, Can. J. Biochem. Physiol., 1961, 39, 1765–1770 CrossRef CAS PubMed.
- H. Shinohara, S. Noguchi, M. Kumazaki, N. Yamada, T. Ito, M. Oyama, Y. Ito, Y. Otsuki, S. Naito and M. Iinuma, J. Toxicol. Res., 2013, 3, 29–34 Search PubMed.
- Y. Shao, Z. Gao, P. A. Marks and X. Jiang, Proc. Natl. Acad. Sci. U. S. A., Early Ed., 2004, 101, 18030–18035 CrossRef CAS PubMed.
- T. Kanzawa, I. Germano, T. Komata, H. Ito, Y. Kondo and S. Kondo, Cell Death Differ., 2004, 11, 448–457 CrossRef CAS PubMed.
- D. Gozuacik and A. Kimchi, Oncogene, 2004, 23, 2891–2906 CrossRef CAS PubMed.
- R. Scherz-Shouval, H. Weidberg, C. Gonen, S. Wilder, Z. Elazar and M. Oren, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18511–18516 CrossRef CAS PubMed.
- Z. J. Yang, C. E. Chee, S. Huang and F. A. Sinicrope, Mol. Cancer Ther., 2011, 10, 1533–1541 CrossRef CAS PubMed.
- P. Codogno and A. Meijer, Cell Death Differ., 2005, 12, 1509–1518 CrossRef CAS PubMed.
- G. Kroemer and B. Levine, Nat. Rev. Mol. Cell Biol., 2008, 9, 1004–1010 CrossRef CAS PubMed.
- N. Dupont, S. Chauhan, J. Arko-Mensah, E. F. Castillo, A. Masedunskas, R. Weigert, H. Robenek, T. Proikas-Cezanne and V. Deretic, Curr. Biol., 2014, 24, 609–620 CrossRef CAS PubMed.
- A. R. Thiam, R. V. Farese Jr and T. C. Walther, Nat. Rev. Mol. Cell Biol., 2013, 14, 775–786 CrossRef CAS PubMed.
- R. Singh and A. M. Cuervo, Int. J. Cell Biol., 2012, 2012, 1–12 CrossRef PubMed.
- R. Singh, S. Kaushik, Y. Wang, Y. Xiang, I. Novak, M. Komatsu, K. Tanaka, A. M. Cuervo and M. J. Czaja, Nature, 2009, 458, 1131–1135 CrossRef CAS PubMed.
- K. Liu and M. Czaja, Cell Death Differ., 2013, 20, 3–11 CrossRef CAS PubMed.
- A. D. Watson, J. Lipid Res., 2006, 47, 2101–2111 CrossRef CAS PubMed.
- M. R. Wenk, Nat. Rev. Drug Discovery, 2005, 4, 594–610 CrossRef CAS PubMed.
- C. Dall'Armi, K. A. Devereaux and G. Di Paolo, Curr. Biol., 2013, 23, R33–R45 CrossRef PubMed.
- M. Haag, A. Schmidt, T. Sachsenheimer and B. Brügger, Metabolites, 2012, 2, 57–76 CrossRef CAS PubMed.
- J. W. Lee, S. Nishiumi, M. Yoshida, E. Fukusaki and T. Bamba, J. Chromatogr. A, 2013, 1279, 98–107 CrossRef CAS PubMed.
- H. J. Mok, H. Shin, J. W. Lee, G.-K. Lee, C. S. Suh, K. P. Kim and H. J. Lim, PLoS One, 2016, 11, e0148577 Search PubMed.
- K. Hsieh, Y. K. Lee, C. Londos, B. M. Raaka, K. T. Dalen and A. R. Kimmel, J. Cell Sci., 2012, 125, 4067–4076 CrossRef CAS PubMed.
- K. Athenstaedt and G. Daum, Cell. Mol. Life Sci., 2006, 63, 1355–1369 CrossRef CAS PubMed.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–39 CrossRef CAS PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- H. Shinohara, K. Taniguchi, M. Kumazaki, N. Yamada, Y. Ito, Y. Otsuki, B. Uno, F. Hayakawa, Y. Minami and T. Naoe, Cancer Lett., 2015, 360, 28–38 CrossRef CAS PubMed.
- S. Mei, H. Gu, A. Ward, X. Yang, H. Guo, K. He, Z. Liu and W. Cao, J. Biol. Chem., 2012, 287, 11761–11768 CrossRef CAS PubMed.
- N. Krahmer, Y. Guo, F. Wilfling, M. Hilger, S. Lingrell, K. Heger, H. W. Newman, M. Schmidt-Supprian, D. E. Vance and M. Mann, Cell Metab., 2011, 14, 504–515 CrossRef CAS PubMed.
- G. Robert, I. B. Sahra, A. Puissant, P. Colosetti, N. Belhacene, P. Gounon, P. Hofman, F. Bost, J.-P. Cassuto and P. Auberger, PLoS One, 2009, 4, e7889 Search PubMed.
- S. H. Tan, G. Shui, J. Zhou, J. J. E. Li, B.-H. Bay, M. R. Wenk and H.-M. Shen, J. Biol. Chem., 2012, 287, 14364–14376 CrossRef CAS PubMed.
- S. Shahnazari, W.-L. Yen, C. L. Birmingham, J. Shiu, A. Namolovan, Y. T. Zheng, K. Nakayama, D. J. Klionsky and J. H. Brumell, Cell Host Microbe, 2010, 8, 137–146 CAS.
- P. Rockenfeller, M. Koska, F. Pietrocola, N. Minois, O. Knittelfelder, V. Sica, J. Franz, D. Carmona-Gutierrez, G. Kroemer and F. Madeo, Cell Death Differ., 2015, 22, 499–508 CrossRef CAS PubMed.
- J. Müller, M. Petković, J. Schiller, K. Arnold, S. Reichl and J. Arnhold, Luminescence, 2002, 17, 141–149 CrossRef PubMed.
- C.-L. Chang, J.-J. Liao, W.-P. Huang and H. Lee, Autophagy, 2007, 3, 268–270 CrossRef CAS PubMed.
- C. Bedia, T. Levade and P. Codogno, Anti-Cancer Agents Med. Chem., 2011, 11, 844–853 CrossRef CAS PubMed.
- S. Pattingre, C. Bauvy, S. Carpentier, T. Levade, B. Levine and P. Codogno, J. Biol. Chem., 2009, 284, 2719–2728 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01965j |
| This journal is © The Royal Society of Chemistry 2016 |