
Photoelectric properties and potential nitro derivatives sensing by a highly luminescent of Zn(ii) and Cd(ii) metal–organic frameworks assembled by the flexible hexapodal ligand, 1,3,5-triazine-2,4,6-triamine hexaacetic acid†
Abstract
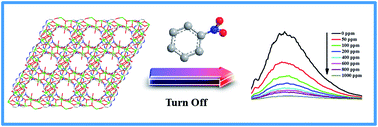
Two new coordination polymers, [Cd4(μ3-O)(TTHA)(H2O)2]·3H2O (1) and [Zn5Na2(TTHA)2(H2O)10] (2), based on the flexible hexapodal ligand 1,3,5-triazine-2,4,6-triamine hexaacetic acid (H6TTHA), have been synthesized under hydrothermal condition. The two coordination polymers were characterized by the elemental analysis, IR spectroscopy, PXRD, thermogravimetric analysis and UV-vis spectroscopy. The single crystal X-ray diffraction analysis revealed that coordination polymer 1 exhibited a 2D sheet structure, which was arranged alternately by two different types of building blocks (Cd4O6 and Cd2O2), and 2 possessed a fascinating 3D network framework with three different types of the building blocks (Zn4(COO)6O8N2, Zn(COO)4 and Na(COO)4O2). In the structures, all of the coordination modes of the ligand were firstly discovered, which are μ8–η1η1η1η3η2η1η2η1η2η1η2 and μ9–η1η1η1η1η1η1η1η1η2ηN1η2η1, respectively. In order to furthermore expand functional characteristics of both the coordination polymers 1 and 2, we also carried out experiments of the surface photovoltage spectroscopy (SPS) and electric-field-induced surface photovoltage spectroscopy (EFISPS), which showed that 1 and 2 could behave as p-type semiconductors. What's more, the fluorescence sensing experiments showed the luminescent quenching of the coordination polymers 1 and 2 were especially obvious with the increasing of the nitro derivatives concentration, even if the nitro derivatives were at a very low concentration, which can also be detected, that indicating both coordination polymers 1 and 2 have very high sensitivity sensing properties.
 Please wait while we load your content...
Please wait while we load your content...

