
Direct access to 2-(hetero)arylated pyridines from 6-substituted 2-bromopyridines via phosphine-free palladium-catalyzed C–H bond arylations: the importance of the C6 substituent†
Abstract
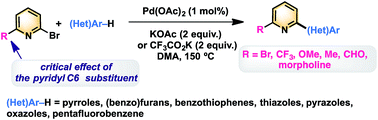
A phosphine-free palladium catalytic system was found to be very active to promote C–H bond activation/arylation of 5-membered ring heterocycles or electron-deficient arenes with 6-substituted 2-bromopyridines, providing a general, straightforward and environmentally benign synthetic approach to a broad variety of 2-(hetero)arylpyridines. The reactivity of 2-bromopyridines is strongly dependent on the substituent at the C6 position. Several C6 substituents such as Br, CF3, CH3, CHO, or morpholine have been employed. Moreover, the use of 2,6-dibromopyridine as the coupling partner allowed the synthesis in high yields of symmetrical and unsymmetrical 2,6-di(hetero)arylpyridines, as well as 2-heteroarylpyridines after the C–Br bond cleavage.
 Please wait while we load your content...
Please wait while we load your content...

