DOI:
10.1039/C6RA01781A
(Paper)
RSC Adv., 2016,
6, 31762-31768
Electrochemistry and structure of Li-rich cathode composites: Li1.26Fe0.22Mn0.52O2in situ integrated with conductive network-graphene oxide for lithium-ion batteries
Received
20th January 2016
, Accepted 21st March 2016
First published on 22nd March 2016
Abstract
A novel designed graphene oxide integrated Li-rich cathode material Li1.26Fe0.22Mn0.52O2 (GO-LFMO) is obtained by integrating nanosized Li1.26Fe0.22Mn0.52O2 (LFMO) materials with different amounts of graphene oxide (GO) through an in situ method. The graphene oxide is designed as the carbon conductive network to regulate and control the micro/nano structure of the composites. Scanning electron microscopy (SEM) reveals that the plate shape composite consists of nano particles with uniform particle size. The elemental mapping results prove that the carbon conductive framework is formed uniformly in the composites. The rate performances of the GO integrated samples are highly improved compared with the pristine one due to the superior conductivity of the conductive networks. Electrochemical Impedance Spectroscopy (EIS) results demonstrate that the graphene oxide conductive networks can remarkably decrease the cell resistance especially the charge transfer resistance.
Introduction
With the growing demand for energy storage in our daily life, lithium-ion batteries (LIBs) are becoming the most promising power supply devices for both small portable electrical appliances and large electric vehicles (EVs).1–4 However, conventional cathode materials such as layered LiCoO2,5,6 olivine LiFePO4 (ref. 7 and 8) and spinel LiMn2O4 (ref. 9 and 10) cannot meet the energy and power density demands. Therefore, novel cathode materials with higher energy density, lower cost, longer cycle life and faster charge/discharge rate are becoming the main research trends.11,12 Since Thackeray's group demonstrated the lithium rich layered oxides (LLOs) materials with capacity of ∼250 mA h g−1 could be the candidates for the cathode materials, the Li rich cathode materials have been widely investigated in recent years.13–20 The Li-rich cathode materials are usually considered as the solid solution of layered Li2MnO3 with C2/m symmetry and layered LiMO2 (M = Ni, Co, Fe, …) with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry.21–24 Among them, the Li rich Fe–Mn based materials Li1+x(FeyMn1−y)1−xO2 have the merits of high capacity (250 mA h g−1), environmental friendly (without cobalt) and low cost (majority of iron and manganese) when compared with Co and Ni.25–28 However, the Li1+x(FeyMn1−y)1−xO2 cathode materials reported so far have generally shown low rate capabilities resulting from the low electronic and ionic conductivities of the material, which greatly limits its practical applications.29,30 Accordingly, to improve the poor electronic and ionic conductivities, much efforts including cation-doping31,32 and the surface modification by coating with carbon33 have been made to enhance the electrochemical performances of Li1+x (FeyMn1−y)1−xO2. However, most of these modified methods cannot improve the electronic and ionic conductivities significantly. Researchers have confirmed that constructing an integrated structure of cathode materials and carbon conductive networks would enhance the electronic conductivity dramatically and thus improve the electrochemical performance.34–38 Therefore, we introduced the conductive network-graphene oxide into LLFO for the first time by an in situ method.
m symmetry.21–24 Among them, the Li rich Fe–Mn based materials Li1+x(FeyMn1−y)1−xO2 have the merits of high capacity (250 mA h g−1), environmental friendly (without cobalt) and low cost (majority of iron and manganese) when compared with Co and Ni.25–28 However, the Li1+x(FeyMn1−y)1−xO2 cathode materials reported so far have generally shown low rate capabilities resulting from the low electronic and ionic conductivities of the material, which greatly limits its practical applications.29,30 Accordingly, to improve the poor electronic and ionic conductivities, much efforts including cation-doping31,32 and the surface modification by coating with carbon33 have been made to enhance the electrochemical performances of Li1+x (FeyMn1−y)1−xO2. However, most of these modified methods cannot improve the electronic and ionic conductivities significantly. Researchers have confirmed that constructing an integrated structure of cathode materials and carbon conductive networks would enhance the electronic conductivity dramatically and thus improve the electrochemical performance.34–38 Therefore, we introduced the conductive network-graphene oxide into LLFO for the first time by an in situ method.
Graphene has large surface area, excellent structural flexibility, superior electrical conductivity and high chemical and thermal stability.39–41 Graphene oxide also shows superior stability and good conductivity. Thus, graphene oxide shows great applicable potential as a conductive additive in Li ion battery, especially used to construct the conductive network. In this work, the conductive network was fabricated using graphene oxide. The graphene oxide integrated Li-rich Fe–Mn based cathode materials were successfully synthesized by an in situ method to obtain the high rate capacity and better cycling stability cathode materials. The conductive network was explored to improve the electrochemical performances especially the rate capability.
Experimental
Materials synthesis
All reagents are analytical grade and used as-purchased without further purification. In a typical procedure, Graphene Oxide (GO) powder was stirred and ultrasonicated with ethanol for at least two hours to obtain the dispersed GO solution (2 mg ml−1). Li1.26Fe0.22Mn0.52O2 (LFMO) were synthesized using molten salt method according to our previous report.33 Stoichiometric amounts of lithium acetate (LiCH3COO·2H2O, mp: 56–58 °C), ferric nitrate (Fe(NO3)3·9H2O, mp: 47.2 °C) and manganese acetate (Mn(CH3COO)2·4H2O, mp: 80 °C) salts were mixed in a beaker with 5 mol% excess of LiCH3COO·2H2O using to compensate any lithium evaporative losses. Then these metal salts were heated at 80 °C in a water bath with mechanical stirring to obtain the well-dispersed mixture. Meanwhile, to prepare the GO-LFMO composite by the in situ method, the dispersed GO solution was added to the molten metal salts drop by drop under continuous stirring in order to mix well. The resulting suspension was stirred at 80 °C in a water bath for 1 h and evaporated to form a gelatinous material. It was then preheated at 200 °C for 10 h and ground into the fine powder. Finally, the material was heated again at 500 °C at a rate of 5 °C min−1 and annealed at that temperature for 5 h under air atmosphere to obtain the GO-LFMO cathode material. The samples with different GO amounts of 1, 2, 3 wt% are labelled as 1% GO-LFMO, 2% GO-LFMO and 3% GO-LFMO. For comparison, the pristine materials were also synthesized without GO under the same condition.
Characterization of the materials
X-ray diffraction (XRD, Bruker, D8 Advance, Germany) was carried out with Cu Kα (40 kV, 40 mA) radiation to identify the crystal structure of all the as-prepared samples at a scanning rate of 0.02° s−1. Raman spectra were measured with a Raman microscopy (HORIBA LabRAM HR Evolution, France) using radiation at 532 nm, and measurements were performed at a laser incident power of 0.3 mW. Special care was taken to avoid the sample damaged by the laser-induced heating. The morphology and element mapping (using copper conductive adhesive) of the as-synthesized powders were characterized using a field emission scanning electron microscopy (FESEM, Hitachi, S-4300 coupled with EDX, Japan) operating in high-vacuum mode. The structure details of the materials were analyzed by the high resolution transmission electron microscope (HRTEM, JEM-2100F, Japan) and energy-dispersive X-ray (EDX) operated at 200 kV. The cathode for HRTEM was dispersed in ethanol, and then was transferred onto microgrids. X-ray photoelectron spectroscopy (XPS, Kratos Analytical Ltd.) was performed with monochromatized Al Kα radiation and an energy resolution of 0.48 eV.
Electrochemical measurements
Electrochemical characterizations were performed in a coin cell (CR2032) assembled with LFMO/GO-LFMO as the cathode and a lithium metal as the anode electrode separated by glass microfiber filters (GF/D, Whatman). The cathode electrode was prepared by coating the slurry of a mixture of 80 wt% active material, 10 wt% super P carbon black and 10 wt% binder (polyvinylidene fluoride) in N-methyl-2-pyrrolidone (NMP) onto Al foil. The electrodes were dried at 80 °C for 6 h and pressed under the pressure of 20 MPa, and then the cathode electrodes were dried at 120 °C for 24 h prior to use. The CR2032 coin cell were assembled in an argon-filled glove box (H2O, O2 < 0.1 ppm). The electrolyte solution was 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. The galvanostatic charge/discharge profiles were measured using a multichannel battery testing system (Neware, Shenzhen, China) at a voltage range of 2.0–4.8 V with 0.2C, 1C, 2C, 3C, 5C (1C = 200 mA g−1) at room temperature. The electrochemical impedance spectroscopy (EIS) of the sample was determined by an electrochemical workstation (VMP3, Biologics, France). The impedance spectra were obtained by applying a sine wave with an amplitude of 5.0 mV over the frequency range of 100 kHz to 10 mHz.
:1 volume ratio. The galvanostatic charge/discharge profiles were measured using a multichannel battery testing system (Neware, Shenzhen, China) at a voltage range of 2.0–4.8 V with 0.2C, 1C, 2C, 3C, 5C (1C = 200 mA g−1) at room temperature. The electrochemical impedance spectroscopy (EIS) of the sample was determined by an electrochemical workstation (VMP3, Biologics, France). The impedance spectra were obtained by applying a sine wave with an amplitude of 5.0 mV over the frequency range of 100 kHz to 10 mHz.
Results and discussion
Crystalline structure analysis
Powder XRD patterns of the pristine and GO modified samples are shown in Fig. 1. All diffraction peaks are sharp and well-defined, suggesting that the prepared compounds are well crystallized and having a pure layer structure. All samples could be well indexed based on the α-NaFeO2 structure (space group Rm), except for the weak superlattice peaks observed between 20° and 22° which correspond to the ordering of Li and Mn in the transition metal layer.27 It indicates the existence of a layered structure with Li2MnO3 character which can be indexed to the monoclinic unit cell C2/m.14,23 The integrated materials do not appear to have any changes in the main reflections and no additional reflections corresponding to graphene are observed due to the low content. The lattice parameters of Rietveld refinements are given in Table 1. Owing to the main peaks of the LFMO are the Rm space group, the high-symmetry Rm space group was employed for the whole pattern refinements rather than the monoclinic unit cell C2/m. Therefore, the superlattice peaks between 20° and 22° were excluded and the accurate refinement results were obtained according to the low R values. The lattice parameters c of the GO-LFMO are larger than the LFMO, which can have wider Li+ extraction and interaction path in the composites. Thus, the better electrochemical performances of the GO integrated LFMO can be predicted from the lattice parameters. The c/a values of all the samples are higher than 4.90 indicating all the samples have the well layered structure. The c/a values are also increased when the GO is integrating with the LFMO. In general, the higher c/a ratio means the better layered structural properties.
 |
| | Fig. 1 XRD patterns of LFMO, 1% GO-LFMO, 2% GO-LFMO and 3% GO-LFMO. | |
Table 1 Crystallographic parameters of Rietveld refinements for LFMO, 1% GO-LFMO, 2% GO-LFMO and 3% GO-LFMO
| Sample |
a (Å) |
c (Å) |
v (Å3) |
c/a |
R
|
| LFMO |
2.878(3) |
14.227(1) |
102.1 |
4.943 |
5.27 |
| 1% GO-LFMO |
2.875(9) |
14.236(5) |
102.0 |
4.952 |
5.53 |
| 2% GO-LFMO |
2.879(5) |
14.236(6) |
102.2 |
4.945 |
5.11 |
| 3% GO-LFMO |
2.878(9) |
14.235(7) |
102.2 |
4.946 |
6.51 |
The Raman spectra of the materials are shown in Fig. 2. The LFMO and GO-LFMO show the same bands at 424, 479 and 592 cm−1. The bands at 479 and 592 cm−1 are attributed to the Eg and A1g vibrations of Raman-active LFMO with the Rm symmetry, respectively. The 424 cm−1 peak is known to be the fingerprint vibration of Li2MnO3.37,42 The absence of the characteristic peak of cubic spinel at 630 cm−1 indicates that the prepared samples are single phase layered materials.43 In the Raman spectrum of GO-LFMO, the peaks observed at 1341 and 1584 cm−1 correspond to the D and G bands of graphene. The D bands stand for the disorder or the defects in the graphite structure, whereas G bands arise from the vibration of sp2 carbon atoms in graphene. These two bands prove that the graphene is successfully integrated with LFMO composite.
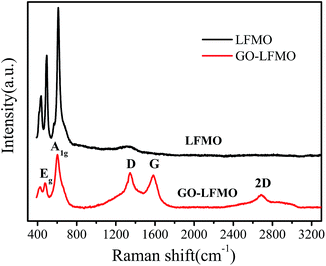 |
| | Fig. 2 Raman spectra of LFMO and GO-LFMO. | |
Morphology analysis
The morphologies of LFMO and GO-LFMO studied by SEM are shown in Fig. 3. All the samples show smooth surface with well-distributed particles. Fig. 3A and B show that the pristine LFMO and the low content of GO (1% GO-LFMO) samples consist of nano-sized materials (less than 100 nm) and having irregular shape. The nanoparticles are aggregated into plate shape (3% GO-LFMO) with the increasing ratio of GO. It is because that the nanoparticles assembled onto a certain adhesive layer which is maybe the GO layer especially in the 3% GO-LFMO (Fig. 3D). Since the amount of GO is extremely low comparing with LFMO, the GO sheets have been fully occupied by the LFMO nanoparticles. The micro/nano structure of GO-LFMO composite is in the sizes of tens of micrometres (Fig. 3D). The morphology is benefit to the improvement of electrochemical behaviour, since the conductivity of the cathode materials can be remarkably improved by the graphene conductive network. Although the GO sheets cannot be observed obviously in the SEM image, it can be proved by the EDX spectra. EDX spectroscopy was employed to investigate the compositions and distribution of different elements in the samples, and the 1% GO-LFMO is chosen to present the results. Fig. 4 reveals that the sample is composed of the expected elements (C, Fe, Mn, O) and the experimental contents of different elements are well consistent with the stoichiometric ratio, as displayed in the table inset the figure, indicating that the sample was synthesized with good stoichiometry. In addition, all the elements disperse uniformly observed from the mapping distribution results.
 |
| | Fig. 3 SEM image of (A) LFMO, (B) 1% GO-LFMO, (C) 2% GO-LFMO, (D) 3% GO-LFMO. | |
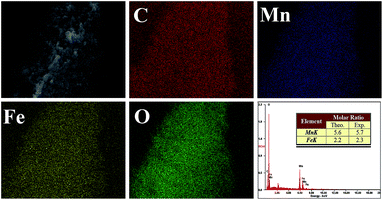 |
| | Fig. 4 EDS and elements distribution of the synthesized sample 1% GO-LFMO. | |
To further understand the GO-LFMO composite in detail, we conducted the TEM analysis of the pristine and the 3% GO-LFMO samples which are shown in Fig. 5. In Fig. 5A and C, the images clearly show the difference of pristine sample and the 3% GO-LFMO sample in morphology. Due to the effect of GO, the nanoparticles of 3% GO-LFMO sample were tightly arrayed and the plate shape were formed. In contrast, without GO, the nanoparticles of pristine material were distributed loosely, without special shape. It indicates that the GO sheets promote the formation of larger and regular particles. From the HRTEM images of the two samples, the distance of lattice fringes were measured to be both 0.48 nm, which are the typical value of layered (003) fringes of Rm. It can be also confirmed that the structure of the bulk LFMO were not affected by the integrating of GO. In addition, the GO acted as a conductive network and connected the nanoparticles of LFMO. After in situ introducing GO into the LFMO, the wide interspaces between the single nano LFMO particles are eliminated. Such an interconnected conductive network forms an efficient electron pathway and thus improves the electronic conductivity of the whole electrode. Furthermore, the tightly arrangement of nano particles on GO can be beneficial for shortening the Li+ diffusion in the electrode. Thus, this kind of micro/nano structure can improve the electrochemical performance of the electrode effectively from the aspect of ionic and electronic transportation. In Fig. 6, the elemental mapping results of the selected area in Fig. 5C obtained from the scanning TEM (STEM) images reveal that the distribution of C, Fe, Mn and O were quite uniform with the same shape of the TEM image. It indicates the carbon conductive networks are successfully introduced into the Li rich Fe–Mn based materials via the in situ method.
 |
| | Fig. 5 (A) TEM image of the pristine sample; (B) HRTEM of the pristine; (C) TEM image of 3% GO-LFMO; (D) HRTEM of 3% GO-LFMO. | |
 |
| | Fig. 6 STEM image and elemental mapping results of 3% GO-LFMO. | |
X-ray photoelectron spectroscopy (XPS) analysis
The pristine and 3% GO-LFMO samples were investigated by the XPS to determine the surface chemical compositions and oxidation state of the elements. Fig. 7a depicts the full spectra of the sample, in which Fe, Mn, O and C species were observed. The Fe 2p3/2 and 2p1/2 exhibit two main peaks with binding energy (BE) of 710.6 and 724.6 eV, respectively, which agrees well with Fe3+.29 Another two peaks at 718.6 and 732.8 eV are satellite peaks, which are also attributed to Fe3+.44Fig. 3C shows the main peaks of Mn 2p3/2 and 2p1/2 are 642.2 and 654.2 eV. These values match well with the binding energy reported for Mn4+ in literature.30,45
 |
| | Fig. 7 XPS spectra of (a) the full spectra, (b) Fe 2p, (c) Mn 2p of LFMO and 3% GO-LFMO (d) C 1s of 3% GO-LFMO. | |
Therefore, the XPS spectra show that the predominant oxidation state of Fe and Mn in the sample is +3 and +4, respectively. After integrating with graphene oxide, the oxidation state of Fe and Mn in the GO-LFMO has not changed.
The C 1s spectrum is shown in Fig. 7d. The C 1s peak at 284.8 eV mainly represents graphitic carbon, which indicates the existence of GO. The binding energy of 289.5 eV corresponds to the adsorption of CO2 in the atmosphere and the formation of Li2CO3 due to the acetate row materials.
Electrochemical performances of pristine and GO-LFMO composite samples
The LFMO and GO-LFMO cathode materials were prepared to electrodes and assembled into coin cells to evaluate their electrochemical performances. Fig. 8 compares the first and second charge/discharge curves and the corresponding cycle performances of LFMO and GO-LFMO at 0.2C and 1C in the range between 2.0 and 4.8 V. For all the samples, Fig. 8a shows a slope below 4.5 V and a plateau above 4.5 V in the first charge profiles, which has the typical characteristics of Li rich cathode materials. The slope below 4.5 V is attributed to the extraction of Li+ from the LiFeO2. The plateau above 4.5 V is attributed to the extraction of Li+ from the Li2MnO3 phase with the form of Li2O. This process is the electrochemical activity of Li2MnO3 phase and a large irreversible capacity loss has formed. From the second charge–discharge process, the plateau around 4.5 V is disappearing and the coulombic efficiency is above 95%. From Fig. 8c, the pristine, 1% GO-LFMO, 2% GO-LFMO and 3% GO-LFMO samples deliver the initial discharge capacity of 229.7, 222.4, 242.5 and 216.4 mA h g−1. The sample 3% GO-LFMO maintains at 134.9 mA h g−1 after 50 cycles with capacity retention of 62.3%. While the pristine sample only has 112.7 mA h g−1 at the 50th cycle with less than 50% capacity retention, which is attributed to the fully exposure to the electrolyte and the side reactions initiated by the electrolyte. The GO in the GO-LFMO composite can prevent some LFMO materials contacting with the electrolyte directly, and the GO conductive networks can improve the electronic conductivity of the cathode. So, the GO-LFMO composite has a much better cycle performance than the LFMO at 0.2C. Similarly trends can also be observed at the rate of 1C in the Fig. 8d. All the samples have the similar initial discharge capacity at the rate of 1C, the 3% GO-LFMO still has the highest capacity (146.7 mA h g−1) and capacity retention (77.71%) at the 50th cycle which are much higher than the pristine sample (107.7 mA h g−1, 57.01%).
 |
| | Fig. 8 The initial and second charge/discharge voltage profiles of all the samples at 0.2C (a) and 1C (b); their cycling performances at 0.2C (c) and 1C (d). | |
To evaluate the rate capability of the materials, the cells were tested from 0.2C to 5C between the voltage limit of 2–4.8 V. As shown in Fig. 9a, the discharge capacity of all the samples is decreasing as the rate increasing. The sample LFMO nearly has no discharge capacity at 3C, while the 3% GO-LFMO delivers a discharge capacity of about 110 mA h g−1; even at 5C, the 3% GO-LFMO sample has a discharge capacity as high as 85 mA h g−1. In addition, the 3% GO-LFMO cathode has the best cycling stability at various rates. The 3% GO-LFMO composite can reach 75% of its initial capacity while the pristine only has 58% of the initial capacity when cycled from 5C to 0.2C, which indicates that the micro/nano structure of the GO-LFMO can enhance the recovery rates capability. The charge/discharge profiles of all the samples at different rates exhibit that the GO-LFMO can decelerate the voltage decay comparing to the LFMO. Thus, the GO-LFMO has much better rate performance than the LFMO. The excellent cycling stability and rate performances of GO-LFMO samples demonstrate that the designed conductive network is effective to improve the electrochemical capability of LFMO.
 |
| | Fig. 9 (a) Rate capability of all the samples at room temperature from the range of 2–4.8 V; (b) their charge/discharge profiles at different rates. | |
To understand the effects of graphene oxide on the electrochemical properties of LFMO, electrochemical impedance spectrums (EIS) were measured for LFMO and GO-LFMO after 50 charge–discharge cycles at 0.2C and the corresponding Nyquist plots are shown in Fig. 10. Before the EIS measurements, all the samples were charged to 4.3 V at a constant current followed by an additional constant voltage charge step. Fig. 10 reveals that all the EIS spectra have two semicircles and a straight sloping line, which can be fitted by the equivalent circuit as shown in the inset of Fig. 10. The first semicircle in the high-frequency region is related to the resistance of surface films (Rs) and the second semicircle can be attributed to the charge transfer resistance (Rct). The slope in the low-frequency region results from lithium ion diffusion in the bulk material. The total resistance of the sample (Rtotal) consists of Rs and Rct which reflects the electrochemical stability and kinetics of electrode reaction.33 As can be seen clearly, the Rct was reduced dramatically comparing with the pristine sample when the LFMO was combined with GO. Especially, the 3% GO-LFMO exhibits a smaller Rct (68 Ω) than other samples (220 Ω, 129 Ω and 111 Ω for the pristine, 1% GO-LFMO and 2% GO-LFMO respectively) since the improvement of electronic conductivity, indicating good reaction kinetics as well as the excellent charge–discharge performance. Therefore, the GO can improve the kinetics of electrode reaction and the electrochemical stability.
 |
| | Fig. 10 The EIS spectra of the pristine and GO integrated samples at charged state of 4.3 V after 50 cycles and the inset figure shows the equivalent circuit for the EIS measurement. | |
Conclusions
In summary, we fabricated a GO conductive network combined Li-rich Fe–Mn based cathode materials by in situ integrating the nanosized LFMO with flaky conductive networks. The rate and cycling tests prove that the micro/nano structure between the micro GO conductive networks and nano LFMO can significantly improve the rate and cycling performances of LFMO. The remarkably improvement of electrochemical performances were achieved by the rapid lithium ion diffusion and high structural stability brought by the micro/nano structure. The electrochemical impedance spectroscopy demonstrates that the dispersed conductive networks in the structure can provide open channels to facilitate the electrolyte flow and soakage, increase the electronic conductivity and decrease the resistance of lithium ion diffusion. Therefore, GO-integrated Li1.26Fe0.22Mn0.52O2 is a promising cathode material in lithium-ion batteries.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21301013).
Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14 CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414 CrossRef CAS PubMed.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091 CrossRef CAS.
- S. Laubach, S. Laubach, P. C. Schmidt, D. Ensling, S. Schmid, W. Jaegermann, A. Thissen, K. Nikolowski and H. Ehrenberg, Phys. Chem. Chem. Phys., 2009, 11, 3278 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- T. F. Liu, J. X. Qiu, B. Wang, Y. Z. Wang, D. L. Wang and S. Q. Zhang, RSC Adv., 2015, 5, 100018 RSC.
- A. Cao and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 6724 RSC.
- J. M. Tarascon, E. Wang, F. K. Shokoohi, W. R. McKinnon and S. Colson, J. Electrochem. Soc., 1991, 138, 2859 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- Y. K. Sun, Z. Chen, H. J. Noh, D. J. Lee, H. G. Jung, Y. Ren, S. Wang, C. S. Yoon, S. T. Myung and K. Amine, Nat. Mater., 2012, 11, 942 CrossRef CAS PubMed.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694 CrossRef CAS PubMed.
- C. H. Shen, S. Y. Shen, F. Fu, C. G. Shi, H. Y. Zhang, M. J. Pierre, H. Su, Q. Wang, B. B. Xu, L. Huang, J. T. Li and S. G. Sun, J. Mater. Chem. A, 2015, 3, 12220 CAS.
- W. K. Pang, S. Kalluri, V. K. Peterson, S. X. Dou and Z. P. Guo, Phys. Chem. Chem. Phys., 2014, 16, 25377 RSC.
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404 CrossRef CAS PubMed.
- Y. J. Zhao, S. J. Wang, W. F. Ren and R. Wu, J. Electrochem. Soc., 2013, 160, A82 CrossRef CAS.
- D. Kim, G. Sandi, J. R. Croy, K. G. Gallagher, S. H. Kang, E. Lee and M. M. Thackeray, J. Electrochem. Soc., 2013, 160, A31 CrossRef CAS.
- Y. J. Zhao, M. H. Xia, X. S. Hu, Z. K. Zhao, Y. Wang and Z. Lv, Electrochim. Acta, 2015, 174, 1167 CrossRef CAS.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614 CrossRef CAS.
- H. Yu, H. Kim, Y. Wang, P. He, D. Asakura, Y. Nakamura and H. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 6584 RSC.
- J. S. Kim, C. S. Johnson, J. T. Vaughey, M. M. Thackeray, W. Hackney and C. P. Grey, Chem. Mater., 2004, 16, 1996 CrossRef CAS.
- J. H. Yan, X. B. Liu and B. Y. Li, RSC Adv., 2014, 4, 63268 RSC.
- M. Tabuchi, H. Shigemura, K. Ado, H. Kobayashi, H. Sakaebe, H. Kageyama and R. Kanno, J. Power Sources, 2001, 97, 415 CrossRef.
- M. Tabuchi, A. Nakashima, H. Shigemura, K. Ado, H. Kobayashi, H. Sakaebe, H. Kageyama, T. Nakamura, M. Kohzaki, A. Hirano and R. Kanno, J. Electrochem. Soc., 2002, 149, A509 CrossRef CAS.
- M. Tabuchi, Y. Nabeshima, T. Takeuchi, K. Tatsumi, J. Imaizumi and Y. Nitta, J. Power Sources, 2010, 195, 834 CrossRef CAS.
- M. Tabuchi, Y. Nabeshima, K. Ado, M. Shikano, H. Kageyama and K. Tatsumi, J. Power Sources, 2007, 174, 554 CrossRef CAS.
- Y. J. Zhao, G. P. Sun and R. Wu, Electrochim. Acta, 2013, 96, 291 CrossRef CAS.
- F. Wu, X. X. Zhang, T. L. Zhao, L. Li, M. Xie and R. J. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 3773 CAS.
- M. Tabuchi, A. Nakashima, K. Ado, H. Sakaebe, H. Kobayashi, H. Kageyama, K. Tatsumi, Y. Kobayashi, S. Seki and A. Yamanaka, J. Power Sources, 2005, 146, 287 CrossRef CAS.
- M. Tabuchi, Y. Nabeshima, T. Takeuchi, H. Kageyama, J. Imaizumi, H. Shibuya and J. Akimoto, J. Power Sources, 2013, 221, 427 CrossRef CAS.
- Y. J. Zhao, Y. C. Sun, Y. Y. Yue, X. S. Hu and M. H. Xia, Electrochim. Acta, 2014, 130, 66 CrossRef CAS.
- B. Luo and L. J. Zhi, Energy Environ. Sci., 2015, 8, 456 CAS.
- C. Ban, Z. Li, Z. Wu, M. J. Kirkham, L. Chen, Y. S. Jung, E. A. Payzant, Y. Yan, M. S. Whittingham and A. C. Dillon, Adv. Energy Mater., 2011, 1, 58 CrossRef CAS.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2011, 3, 2966 CAS.
- K. Jiang, X. Wu, Y. Yin, J. Lee, J. Kim and Y. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 4858 CAS.
- I. T. Kim, J. C. Knight, H. Celioa and A. Manthiram, J. Mater. Chem. A, 2014, 2, 8696 CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- F. Ding, H. Ji, Y. Chen, A. Herklotz, K. Dörr, Y. Mei, A. Rastelli and O. G. Schmidt, Nano Lett., 2010, 10, 3453 CrossRef CAS PubMed.
- J. R. He, Y. F. Chen, P. J. Li, Z. G. Wang, F. Qi and J. B. Liu, RSC Adv., 2014, 4, 2568 RSC.
- M. Inaba, Y. Iriyama, Z. Ogumi, Y. Todzuka and A. Tasaka, J. Raman Spectrosc., 1997, 28, 613 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441 CrossRef CAS.
- A. Abouimrane, O. C. Cpmpton, H. Deng, I. Belharouak, D. A. Dikin, S. T. Nguyen and K. Amine, Electrochem. Solid-State Lett., 2011, 14, A126 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









