
Facile synthesis and evaluation of quercetin reduced and dextran sulphate stabilized gold nanoparticles decorated with folic acid for active targeting against breast cancer†
Abstract
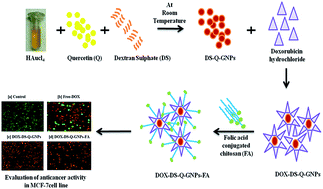
In the present work, gold nanoparticles (GNPs) were successfully prepared by green synthesis using a strong antioxidant Quercetin (Q) as a reducing agent in the presence of Dextran Sulphate (DS) as a stabilizing agent at room temperature (DS–Q–GNPs). DS–Q–GNPs were characterized by several in vitro techniques to understand their physiochemical and biological properties. However, the average particle size was found to be around 38 nm, and the zeta potential of DS–Q–GNPs was found to be around −42 mV, indicating that the particles were highly stable. TEM results showed that the particles prepared were nearly spherical and crystalline in nature. DS–Q–GNPs exhibited good stability and showed excellent biocompatibility in MTT assay using the NIH 3T3 fibroblast cell line. Similarly, hemocompatibility and in vivo zebra fish toxicity studies confirmed that the DS–Q–GNPs were highly biocompatible and safe in vivo. Doxorubicin (DOX) was successfully loaded on DS–Q–GNPs and decorated with folic acid (FA) for targeting drug delivery to the cancer site (DOX–DS–Q–GNPs–FA). In vitro drug release studies revealed the drug was released in a controlled manner. The anticancer activity of DOX–DS–Q–GNPs–FA against the human breast cancer cell lines (MCF-7) was evaluated using MTT assay. Both doxorubicin loaded GNPs and the free doxorubicin inhibit the growth of MCF-7 cells in a concentration-dependant manner. The enhanced activity of DOX–DS–Q–GNPs–FA against MCF-7 cells was observed with its effect on the cell cycle progression of MCF-7 cells using flow cytometry. Similarly, Western blotting was employed to understand the cell cycle pathways during the treatment of DOX–DS–Q–GNPs–FA. It was found that DOX-loaded DS–Q–GNPs conjugated with FA represent a new potential delivery system for improved cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

