
Isolation, synthesis, and biological activity of tomentosenol A from the leaves of Rhodomyrtus tomentosa†
Abstract
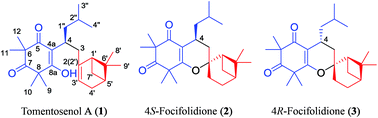
Tomentosenol A (1), along with a pair of epimers, 4S-focifolidione (2) and 4R-focifolidione (3), were isolated from the leaves of Rhodomyrtus tomentosa. 1 was the first example of a new meroterpenoid class with a unique skeleton that contained a free syncarpic acid coupled with a terpenoid unit, and showed excellent antimicrobial and cytotoxic activities. The absolute configuration of 1 was unambiguously determined by a chemical conversion between 1 and the predetermined 2. In contrast to the common hetero Diels–Alder cycloaddition embodied in previously reported meroterpenoid biosynthesis, an Alder-ene reaction was proposed as the key transformation to account for the biosynthesis of 1, which was confirmed by a biomimetic total synthesis.
 Please wait while we load your content...
Please wait while we load your content...

