
3,6-Di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) mediated metal-free mild oxidation of thiols to disulfides in aqueous medium†
Suvendu Samanta,
Shounak Ray,
Abhisek Brata Ghosh and
Papu Biswas*
Department of Chemistry, Indian Institute of Engineering Science and Technology, Shibpur, Howrah, 711 103, India. E-mail: papubiswas_besus@yahoo.com
First published on 14th April 2016
Abstract
Thiols are efficiently oxidized to disulfides (RSSR) in the presence of 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) in aqueous medium, as well as in the absence of a solvent under mild and metal-free conditions. A broad range of alkyl, aryl and heterocyclic symmetrical disulfides can be easily obtained in almost quantitative yields. The X-ray single crystal structure of 2-aminocyclopent-1-ene-1-carbothioic dithioperoxyanhydride (disulfide obtained from 2-aminocyclopentene-1-dithiocarboxylic acid, ACDA) is reported. The reaction mechanism has been studied thoroughly. It is shown that the reaction proceeds through the formation of an organosulphur radical. Pytz interacts with thiol to accept one electron and produces an organosulphur radical. Pytz ultimately accepts two electrons to form H2pytz and is capable of oxidizing 2 equivalents of thiols.
1. Introduction
Oxidative coupling of thiols to disulfides is one of the most important chemical processes in both chemistry and biology. In nature, the formation of disulfide bonds is essential for stabilization of biologically active folded conformations of peptides and proteins.1 Furthermore, control of several metabolic pathways as well as gene expression are related to the enzymatic reduction of disulfide bonds.2 The formation of the disulfide bonds is not only important for living cells, but it also has applications in the chemical industry in anti-oxidants, pharmaceuticals, pesticides and as vulcanization agents.3 Disulfides are also crucial intermediates in the synthesis of sulphur containing heterocycles4 and organic compounds such as sulfonyl, sulfenyl.5 Moreover, due to relative high stability of the disulfide bonds toward organic reactions, such as oxidation, alkylation and acylation, compared to the corresponding free thiols, the protection of thiol groups can conveniently be achieved as disulfide. Consequently, several effective reagents and methods have been developed for the oxidative coupling of thiols, including stoichiometric oxidizing agents such as KMnO4,6a MnO2,6b anhydrous K3PO4,6c dichromates,6d VO(acac)2,6e copper salt,6f rhenium–sulfoxide complex,6g rhodium(I) complex,6h pyridinium chlorochromate,6i pyridinium fluorochromate,6j N-phenyltriazolinedione,6k molecular bromine supported on silica gel,6l trichloroisocyanuric acid,6m 1,3-dibromo-5,5-dimethylhydantoin,6n and diaryl organo telluride.6p–r However, most of these reagents are toxic, hazardous and generate harmful wastes. Numerous homogenous as well as heterogeneous catalysts have also been reported using molecular oxygen as the primary oxidant which include Fe(BTC) MOF (BTC: 1,3,5-benzenetricarboxylate),7a gold nanoparticles deposited on CeO2,7b diaryl tellurides under photosensitized conditions,7c Ag2O nanoparticle incorporated mesoporous silica,7d Eosin Y under visible light irradiation,7e cobalt(II) phthalocyanines in ionic liquid,7f selenium ionic liquid,7g basic and neutral alumina,7h–j iron7k and cobalt7l,m complexes, intercalated [MoVIO2(O2CC(S)Ph2)2]2 in a Zn(II)–Al(III) layered double hydroxide host7n and Ni-nanoparticles.7o The oxidative dimerization of thiols in presence of oxygen was also reported using laccases as catalyst,8a with Et3N in DMF under sonication.8b Catalyst and additive free synthesis of disulfides from thiols in bio-based green solvent ethyl lactate8c and in ethanol have also been reported very recently.8dMost of these catalytic procedures listed above suffer from one or more of the following disadvantages, long reaction times, difficult work-up procedure, require higher or lower temperature to obtain satisfactory results, strong basic or acidic media and require metal based catalyst, sometime relatively expensive metal. Additionally, a large variety of oxidation products can possibly be formed during thiol oxidation, such as sulfones, sulfoxides and sulfonic acids along with disulfide. It is then of interest to develop a mild, efficient, metal free selective process for the transformation of thiols into disulfides avoiding over oxidation to oxygenated sulfur products.
s-Tetrazine derivatives are highly electroactive heterocycles as they have very high electron affinity which makes them easily reducible and the electron poorest C–N heterocycles.9 s-Tetrazine molecules can reversibly be reduced in organic solvents. Almost all tetrazine molecules accept one electron to form a stable anion radical. Tetrazine derivatives can also accept a second electron though addition of second electron is not electrochemically reversible in standard conditions. The corresponding dianion radical formed probably highly basic in character and easily be protonated to produce 1,4-dihydrotetrazine9,10 as shown in Scheme 1a. Furthermore, in aqueous medium, s-tetrazines accepts two electrons and two protons to produce 1,4-dihydrotetrazines9b (Scheme 1b).
 | ||
| Scheme 1 Reduction of s-tetrazine (Tz) to dihydrotetrazine (H2Tz) (a) via anion radical in organic solvent (b) in water. | ||
Interaction of tetrazine molecules with proton donors such as dialkyl thiourea11a and phenol derivative11b has been demonstrated previously. Reduction of 3,6-diphenyl-s-tetrazine (PhTz) by 10-methyl-9,10-dihydroacridine (AcrH2) in presence of scandium ion as promoter has also been reported by Fukuzumi et al.12 In presence of Sc3+, hydride transfer from AcrH2 to PhTz occurs easily at ambient temperature to give 10-methylacridinium ion (AcrH+). So, due to this highly electron deficient nature of s-tetrazines, they can be utilized as two electron oxidising agent under suitable conditions. Recently, we have reported the synthesis of 2-substituted benzimidazoles and benzothiazoles from different aldehydes and oxidation of alcohols to carbonyls using 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) molecule as catalyst under visible light irradiation at ambient temperature.13 In our continuous effort to expand the synthetic utility of pytz molecule (Fig. 1), herein we report the pytz mediated metal-free efficient oxidative coupling of thiols to disulfides under mild conditions in aqueous medium.
 | ||
| Fig. 1 3,6-Di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz). | ||
2. Results and discussion
2.1. Optimization of the reaction conditions
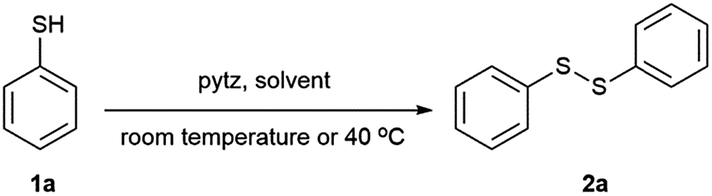
Our initial efforts focused on identifying the optimal reaction conditions for our proposed oxidation and are shown in Table 1. In order to carry out this synthetic protocol, a model reaction of thiophenol (1a) in presence of 1 equivalent 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) in water at room temperature was performed and thiophenol (1a) was smoothly and rapidly converted into diphenyl disulfide (2a) in almost quantitative yield within a very short reaction time (Table 1, entry 1). Besides water, a relatively lower yield was obtained when the reaction was carried out in other organic solvents such as chloroform, dichloromethane, ethanol, methanol, tetrahydrofuran and acetonitrile (Table 1, entries 2–7). Interestingly, desired product was also achieved in quantitative yield when reaction was conducted in absence of any solvent (Table 1, entry 8). The temperature was considered as another possible variable for this method. The reaction time was reduced considerably when the reaction temperature was slightly increased from room temperature to 40 °C (Table 1, entry 9). However, no improvement appeared by further increasing the temperature and lower reaction temperature than room temperature had a negative effect on the reaction yield. Notably, decreasing the amount of pytz to 0.5 equiv. gave the identical result to those of 1.0 equiv. pytz (Table 1, entry 10). The reaction was also carried out in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) water:ethanol mixture anticipating relatively poor solubility of few aromatic thiols in water to get almost quantitative yield (Table 1, entry 11). However, no expected oxidation of thiols occurred without pytz as the oxidant (Table 1, entry 12). In order to exclude the possibility of air being involved in the reaction, a control reaction under an inert atmosphere was performed and no influence on the yield was observed. So, the optimum conditions are reported in entries 10 and 11, whereby water or water:ethanol (9:1) solution of the thiophenol (1 equiv.) and pytz (0.5 equiv.) was stirred at 40 °C. The progress of the reaction was monitored by TLC and gas chromatography and complete conversion was achieved in 12 min. Chromatographic purification of the reaction mixture gave diphenyl disulfide in >98% yield.
:1 (v/v) water:ethanol mixture anticipating relatively poor solubility of few aromatic thiols in water to get almost quantitative yield (Table 1, entry 11). However, no expected oxidation of thiols occurred without pytz as the oxidant (Table 1, entry 12). In order to exclude the possibility of air being involved in the reaction, a control reaction under an inert atmosphere was performed and no influence on the yield was observed. So, the optimum conditions are reported in entries 10 and 11, whereby water or water:ethanol (9:1) solution of the thiophenol (1 equiv.) and pytz (0.5 equiv.) was stirred at 40 °C. The progress of the reaction was monitored by TLC and gas chromatography and complete conversion was achieved in 12 min. Chromatographic purification of the reaction mixture gave diphenyl disulfide in >98% yield.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (min) | Yieldb |
| a Reaction condition (until otherwise specified): 2 mmol thiophenol, solvent (10 mL solvents used for all cases), pytz (1 equiv., 2 mmol).b Isolated yield after chromatographic purification.c Room temperature.d Reaction carried out in presence of 0.5 equiv. (1 mmol) pytz.e Reaction done in absence of pytz. | ||||
| 1 | H2O | RTc | 20 | >98% |
| 2 | CHCl3 | RT | 20 | 91 |
| 3 | CH2Cl2 | RT | 20 | 94 |
| 4 | EtOH | RT | 20 | 97 |
| 5 | MeOH | RT | 20 | 96 |
| 6 | THF | RT | 20 | 89 |
| 7 | CH3CN | RT | 20 | 96 |
| 8 | None | RT | 20 | >98% |
| 9 | H2O | 40 | 12 | >98% |
| 10d | H2O | 40 | 12 | >98% |
| 11d | H2O:EtOH |
40 | 12 | >98% |
| 12e | H2O | 40 | 20 | Trace |
2.2. Scope of the reaction
With the results of our initial feasibility and optimization study in hand, we turned our attention toward exploring the substrate scope of our oxidation protocol. As shown in Scheme 2, the present protocol was found to be advantageous for the oxidation of various aromatic and aliphatic thiols to the corresponding disulfides. All reactions were carried out at 40 °C as well as room temperature. The oxidation of thiophenol and its substituted derivatives to the corresponding disulfides were obtained in almost quantitative yields (Scheme 2, 2a–2f) with no apparent substituent effects. Even, sterically hindered thiophenols bearing ortho carboxylic or amido substituents could efficiently be converted to the corresponding disulfides (Scheme 2, 2e and 2f). Heterocyclic thiols, such as 2-mercapto quinoline (1g), 2-mercaptopyridine (1h), 2-mercapto pyrimidine (1i), 2-mercaptobenzimidazole (1j) and 2-mercaptobenzothiazole (1k), can also be oxidized conveniently to the corresponding disulfides (Scheme 2, 2g–2k). Benzothioic S-acid (1l) and phenylmethanethiol (1m) were smoothly oxidized to give the disulfides in quantitative yields (Scheme 2, 2l and 2m). | ||
| Scheme 2 Oxidation of thiols to disulfides in aqueous medium. Reaction conditions (until otherwise specified): thiols (2 mmol), pytz (1 mmol), 9:1H2O/EtOH (10 mL), 40 °C or room temperature. Isolated yields are shown. Time required for room temperature reaction is shown in parenthesis. aReaction carried out in water only. | ||
The scope of this oxidation methodology was further expanded using aliphatic thiols (Scheme 2, 2n–2q). Our protocol demonstrated selective oxidation of the thiol in the presence of hydroxy functional group (Scheme 2, 2q). Ethanethioic S-acid (1r) was also effectively oxidized to the corresponding disulfide (Scheme 2, 2r).
Conversion of the biologically important cysteine into the cystine was also investigated. Clean and quantitative formation of cystine in water was observed within a very short reaction time (Scheme 2, 2s). In our study, the reaction time required for above conversion is lower than that of previously reported protocols (which are typically varies from one to few hours for other recently published protocols).7b,c,e
Encouraged by these results, we turned our attention to the synthesis of tetraalkylthiuram disulfides. Tetraalkylthiuram disulfides are known for their extensively used in the rubber industry as an essential vulcanization reagent.7e Accordingly, 2-aminocyclopentene-1-dithiocarboxylic acid (ACDA) was oxidized to corresponding disulfide (Scheme 2, 2t). Oxidation of diethylcarbamodithioic acid to disulfiram (Scheme 2, 2u) has also been carried out successfully. Disulfiram, which is commonly sold under the trade name Antabuse, displays biological properties and known drug for treatment of chronic alcoholism.7e,14
In order to test the synthetic utility of this method, 2s was prepared on a gram scale using water as solvent. The oxidation of 1s (10 mmol, 1.21 g) with pytz (5 mmol, 1.18 g) under the optimal reaction conditions afforded 2s in 96% yield (1.15 g).
As was previously observed that the transformation of compound 1a into disulfide 2a also takes place rapidly and efficiently when carried out in absence of solvent (entry 8, Table 1). Therefore, various liquid aryl and alkyl thiols were reacted with 0.5 equivalent pytz at 40 °C as well as ambient temperature in absence of any solvent. Under these reaction conditions, both aromatic and aliphatic liquid thiols were selectively oxidized to their corresponding disulfides in near quantitative yields (Scheme 3).
 | ||
| Scheme 3 Oxidation of liquid thiols to disulfides in absence of solvent. Reaction conditions (until otherwise specified): thiols (2 mmol), pytz (1 mmol), 40 °C or room temperature. Isolated yields are shown. Time required for room temperature reaction is shown in parenthesis. | ||
2.3. Crystal structure
In general, the formation of disulfide (S–S) bond from the oxidation of thiol bond (S–H) could be confirmed from the FT-IR spectrum. To prove formation of disulfide bond unambiguously, single crystal X-ray structure of compound 2t (Scheme 2) has been determined. Crystallographic data and selected details of structure determinations are given in Table S1 (ESI†) and Fig. 2 shows the ORTEP representation of compound 2t. The thioperoxyanhydride group exhibits an open book structure. The S–S bond distance is found to be 2.0112(7) Å and the dihedral angle between two S–S–C planes (S2–S3–C7 and S3–S2–C6) is 87.99°. The two C–S bond distances [1.8069(19) Å for S2–C6 and 1.793(2) Å for S3–C7] are almost identical, as are the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond distances [1.671(2) Å for S1–C6 and 1.669(2) Å for S4–C7]. Two S–S–C bond angles, S3–S2–C6 and S2–S3–C7, are 107.28(7) and 106.41(7), respectively. Both the double bonded sulphur atoms (S1 and S4) form strong intramolecular hydrogen bond with N–H hydrogens. The pertinent bond distances and angles are shown in Table S2 (ESI†).
S bond distances [1.671(2) Å for S1–C6 and 1.669(2) Å for S4–C7]. Two S–S–C bond angles, S3–S2–C6 and S2–S3–C7, are 107.28(7) and 106.41(7), respectively. Both the double bonded sulphur atoms (S1 and S4) form strong intramolecular hydrogen bond with N–H hydrogens. The pertinent bond distances and angles are shown in Table S2 (ESI†).
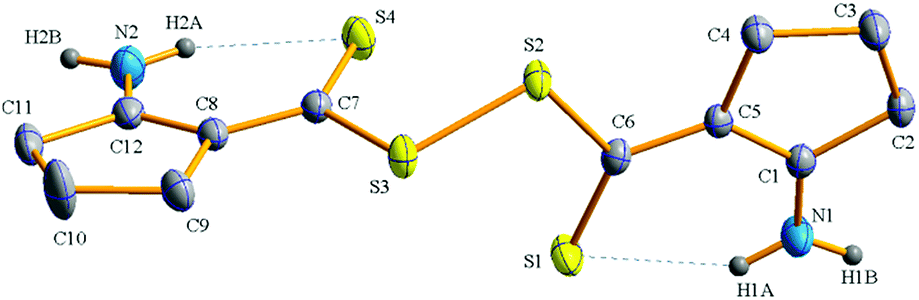 | ||
| Fig. 2 ORTEP representation of the compound 2t showing 50% probability displacement ellipsoids. Hydrogen atoms are omitted for clarity. | ||
2.4. Reaction mechanism
In order to elucidate the mechanism of the pytz mediated oxidation of the thiols to corresponding disulfides, the electrochemical behaviour of the pytz molecule was studied by cyclic voltammetry in water. Pytz shows one quasi-reversible reduction peak at +0.045 V (ΔEp = 0.12 V) in water (Fig. 3). Pytz molecule accepts two electrons and two protons in water to produce H2pytz as demonstrated by Cosnier et al. recently.9d The result is consistent with the fact that tetrazines are chemically reduced by a two-protons, two-electrons process into 1,4-dihydrotetrazines. Based upon these results, a plausible catalytic pathway is proposed in Scheme 4. In presence of two equivalent of thiols, pytz is transformed in to H2pytz by accepting two electrons and two proton from two thiol molecules. On the other hand, thiol (RSH) forms RS˙ radical. Coupling of two RS˙ radical produce RSSR. The formation of H2pytz can be monitored by absorption spectroscopy. Pytz molecule exhibits an absorption maximum at 534 nm in water, whereas H2pytz shows an ill-defined shoulder at about 405 nm in visible region. Accordingly, a water–ethanol (9:1 v/v) solution of pytz (1 mmol) was reacted with thiophenol (2 mmol) and the progress of the reaction was monitored spectrophotometrically. Fig. 4 shows that while the absorption curves pass through the isosbestic points at 473 and 595 nm, the more intense peak at 534 nm gradually disappears at the expense of evolution of a less intense ill-defined shoulder at 405 nm. Furthermore, H2pytz molecule could easily be recovered from the reaction mixture by column chromatography. Recovery of H2pytz was further confirmed by its proton NMR and single crystal X-ray structure (Fig. S1, ESI†). Another interesting aspect of this reaction is that recovered H2pytz could easily be oxidized to pytz with NaNO2/HCl mixture to reuse as recoverable reagent for oxidation of thiols to disulfides.
 | ||
| Fig. 3 Cyclic voltammogram (CV) of 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) in water. Scan rate for cyclic voltammetric measurements were 50 mV s−1. | ||
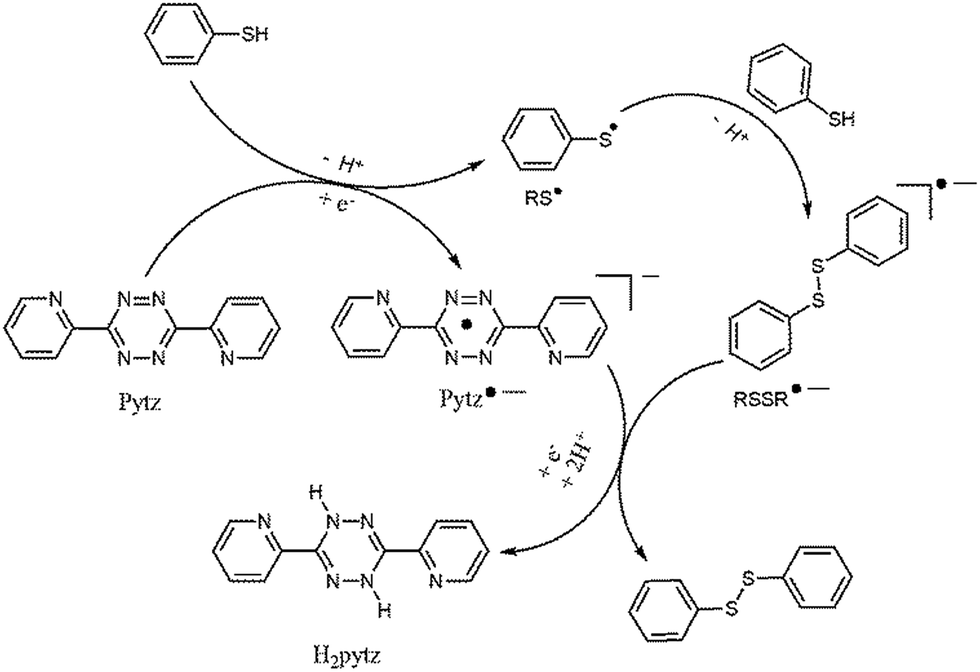 | ||
| Scheme 4 Proposed mechanism for 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) mediated oxidation of thiols to disulfides. | ||
 | ||
| Fig. 4 Spectral changes of 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz, 1 mmol) in presence of thiophenol (2 mmol) in water–ethanol (9:1, v/v) solution. | ||
To prove that the reaction follows a radical pathway, reaction was carried out in the presence of a radical scavenger, TEMPO (2,2,6,6-tetramethylpiperidinoxyl). Addition of TEMPO in a 1:1 molar proportion (with respect to pytz) to the reaction mixture, only trace yield can be identified by GC.
3. Conclusions
In summary, we have developed a highly efficient metal free oxidation of thiols to disulfides using 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) as oxidant. A wide variety of aromatic, aliphatic and heterocyclic thiophenols could be converted in excellent yields under mild conditions to the corresponding disulfides. The present procedure shows the following favourable features: (1) the reaction does not require any metal based oxidant; (2) the reaction proceeds under neutral and mild conditions (neither acid nor base required) and thus making it suitable with acid- or base sensitive substrates; (3) the reaction can be carried out in aqueous medium as well as in the absence of solvents at room temperature; (4) the present method is effective for a wide range of substrates bearing various functional groups; (5) our developed protocol should be suitable for large-scale synthesis of disulfides from thiols. Furthermore, H2pytz could be recovered from the reaction mixture, which can be re-oxidized by known oxidation procedures to pytz for re-used.4. Experimental
4.1. Materials
Solvents were purified according to standard methods prior to use, while all other substances and reagents used, were commercially available and used as received. The synthesis of 3,6-di(pyridin-2-yl)-1,2,4,5-s-tetrazine (pytz) was carried out following the method reported earlier.9,154.2. Physical measurements
The electronic absorption spectra were measured at room temperature on an Agilent 8453 diode array spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance DPX 300, 400 and 500 MHz NMR spectrometer and 13C NMR spectra were measured using 125 MHz spectrometer. All 1H data were reported in parts per million (ppm) relative to tetramethylsilane (δH = 0) in the deuterated solvents. The electrospray ionization mass spectra (ESI-MS) were measured on a Micromass Qtof YA 263 mass spectrometer. Electrochemical measurements were carried out in water at 25 °C under a nitrogen atmosphere using μAUTOLABIII/FRA2 electrochemical analyzer. Cyclic voltammetric (CV) measurements were carried out with a three-electrode assembly comprising glassy carbon working electrode, a platinum auxiliary electrode, and an aqueous Ag/AgCl reference electrode. The concentration of the supporting electrolyte potassium chloride (KCl) was 0.1 M, while that of the pytz was 1 mM. GC analyses were performed on an Agilent Technologies 6890 N network GC system with an FID detector using a J & W HP-5 column (30 m, 0.32 mm internal diameter) and n-decane as the internal standard. Thin layer chromatography (TLC) was performed on EMD pre-coated plates (silica gel 60 F254, Art 5715) and visualized by fluorescence quenching under UV light. Column chromatography was performed on Silica Gel (60–120 Mesh).4.3. General experimental procedure for the synthesis of disulfide in water ethanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1, v/v)
:1, v/v)
To a solution of thiol (2 mmol) in 10 mL water–ethanol (9:1) was added pytz (1 mmol) and the mixture was stirred at 40 °C or room temperature. The progress of the reaction was monitored by TLC and gas chromatography. After completion of the reaction, solvent was evaporated at reduced pressure and the product was dissolved in minimum volume of ethyl acetate. The solvent was concentrated in vacuo, and purified by column chromatography using silica gel (hexane/ethyl acetate) to get desired product.
4.4. General experimental procedure for the synthesis of disulfide in water
To a solution of thiol (2 mmol) in water (10 mL) was added pytz (1 mmol) and the mixture was stirred at 40 °C or room temperature. A white precipitation appeared while stirring. The product was filtered, washed thoroughly with water and dried overnight.:1) following the general method and the product was purified by column chromatography. White solid, yield: 0.21 g, 98%. Mp: 60 °C. 1H NMR (400 MHz, CDCl3, δH ppm): 7.504 (d, 4H, J = 7.6), 7.306 (t, 3H, J = 7.6, 8.0), 7.243 (t, 3H, J = 7.2, 7.6).:1) following the general method and the product was purified by column chromatography. Off yellow liquid, yield: 0.27 g, 98%. 1H NMR (400 MHz, CDCl3, δH ppm): 7.388 (m, 4H), 6.819 (m, 4H), 3.794 (s, 6H).:1) following the general method and the product was purified by column chromatography. Yellow solid, yield: 0.29 g, 98%. 1H NMR (400 MHz, CDCl3, δH ppm): 7.891 (d, 4H, J = 8.0), 7.541 (d, 4H, J = 8.0), 2.544 (s, 6H).:1) following the general method and the product was purified by column chromatography. Yellow solid, yield: 0.24 g, 98%. Mp: 93 °C. 1H NMR (400 MHz, CDCl3, δH ppm): 6.582 (t, 2H, J = 7.6), 6.706 (d, 2H, J = 8.4), 7.152 (dd, 4H, J = 2, 2.4, 3.2), 4.340 (br, 4H).:1) following the general method. White solid precipitated, filtered, washed with water and dried. White solid, yield: 0.30 g, 98%. Mp: 289 °C. 1H NMR (400 MHz, DMSO-d6, δH ppm): 13.538 (s, 2H), 8.030 (dd, 2H, J = 1.2, 0.8), 7.618–7.527 (m, 4H), 7.347–7.307 (m, 2H).:1) following the general method and the product was purified by column chromatography. Off white solid, yield: 0.45 g, 98%. 1H NMR (300 MHz, CDCl3, δH ppm): 8.865 (s, 2H), 8.427 (d, 2H, J = 8.4), 7.616 (t, 4H, J = 6.9, 1.5), 7.513–7.344 (m, 2H), 7.431–7.166 (m, 6H), 7.261–7.189 (m, 2H), 6.872 (dd, 2H, J = 0.9, 6.6, 7.5). 13C NMR (125 MHz, DMSO-d6) δC 167.3, 132.7, 130.7, 129.2, 129.9, 128.5. HRMS (ESI+): m/z = 457.1423, [100%, MH+]; calcd. for C26H21N2O2S2: 457.1044.:1) following the general method and the product was purified by column chromatography. Yellow solid, yield: 0.31 g, 98%. 1H NMR (400 MHz, CDCl3, δH ppm): 7.930 (m, 4H), 7.750 (d, 2H, J = 8), 7.630 (m, 4H), 7.430 (t, 2H, J = 8).:1) following the general method and the product was purified by column chromatography. Yellow solid, yield: 0.21 g, 98%. Mp: 57 °C. 1H NMR (300 MHz, CDCl3, δH ppm): 8.401 (d, 2H, J = 3.9), 7.565 (d, 4H, J = 3.6), 7.071 (t, 2H, J = 3.9, 4.2).:1) following the general method and the product was purified by column chromatography. Yellow solid. yield: 0.22 g, 98%. Mp: 58 °C. 1H NMR (300 MHz CDCl3, δH ppm): 8.572 (dd, 4H, J = 1.2, 3.3), 7.374–7.324 (m, 2H).:1) following the general method and white solid is precipitated, filtered, washed with water and dried. White solid, yield: 0.29 g, 98%. 1H NMR (400 MHz DMSO-d6, δH ppm): 7.719 (dd, 4H, J = 3.2, 2.8), 7.374 (dd, 4H, J = 3.2, 2.8), 6.637 (br, 2H).:1) following the general method and yellow solid is precipitated, filtered, washed with water and dried. Yellow solid, yield: 0.32 g, 98%. Mp: 186 °C. 1H NMR (400 MHz CDCl3, δH ppm): 7.940 (d, 2H, J = 16.0), 7.777 (d, 2H, J = 15.6), 7.510–7.314 (m, 4H).:1) following the general method and the product was purified by column chromatography. White solid, yield: 0.26 g, 98%. Mp: 233 °C. 1H NMR (400 MHz CDCl3, δH ppm): 8.123–8.075 (m, 2H), 7.949–7.925 (m, 1H), 7.683–7.286 (m, 7H).:1) following the general method and the product was purified by column chromatography. White solid, yield: 0.24 g, 98%. Mp: 108–110 °C. 1H NMR (400 MHz CDCl3, δH ppm): 7.349–7.240 (m, 10H), 3.612 (s, 4H).:1) following the general method and the product was purified by column chromatography. Colorless oil, yield: 0.09 g, 98%. 1H NMR (400 MHz, CDCl3, δH ppm): 2.672 (t, 4H, J = 7.6, 7.2), 1.701–1.583 (m, 8H), 1.390–1.296 (m, 28H), 0.876 (t, 6H, J = 6.8).:1) following the general method and yellow solid is precipitated, filtered, washed with water and dried. Off white solid yield: 0.29 g, 99%. 1H NMR (400 MHz, CDCl3, δH ppm): 3.890–3.720 (m, 2H), 2.907–2.777 (m, 2H), 2.695 (dd, 2H, J = 4.4, 16). 13C NMR (125 MHz, CDCl3) δC 179.9, 179.2, 53.1, 40.8. HRMS (ESI+): m/z = 299.1034, [100%, MH+]; calcd for C8H11O8S2: 298.9895.:1) following the general method and the product was purified by column chromatography. Yellow oil. Yield: 0.15 g, 98%. 1H NMR (300 MHz, CDCl3, δH ppm): 3.827 (dd, 4H, J = 5.6, 5.4, 6.6), 2.882–2.796 (m, 4H), 1.993 (t, 2H, J = 7.8, 6.6).:1) following the general method and the product was purified by column chromatography. Off white solid, yield: 0.29 g, 98% mp: 71 °C. 1H NMR (300 MHz, CDCl3, δH ppm): 3.951 (t, 8H, J = 6.0), 1.325 (t, 6H, J = 1.0), 1.235 (d, 6H, J = 5.4). HRMS (ESI+): m/z = 297.0983, [100%, MH+]; calcd for C10H21N2S4: 297.0588.4.5. General experimental procedure for the synthesis of disulfide without solvent
Pytz (1 mmol) was added to the liquid thiol (2 mmol) and the mixture was stirred at 40 °C or room temperature. The progress of the reaction was monitored by TLC and gas chromatography. After completion of the reaction, the mixture was dissolved in minimum volume of ethyl acetate. The solvent was concentrated in vacuo, and purified by column chromatography using silica gel (hexane/ethyl acetate) to get desired product.4.6. X-ray crystallography
Crystals suitable for structure determinations were obtained by slow evaporation of their dichloromethane–acetonitrile solutions. Single crystals were mounted on glass fibers and coated with perfluoropolyether oil. Intensity data were collected at 150(2) K on a Bruker-AXS SMART APEX II diffractometer equipped with a CCD detector using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The data were processed with SAINT,19 and absorption corrections were made with SADABS.19 The structures were solved by direct and Fourier methods and refined by full-matrix least-squares based on F2 using SHELXTL20 and SHELX-97 software packages.21 The non-hydrogen atoms were refined anisotropically, while the hydrogen atoms were placed at geometrically calculated positions with fixed isotropic thermal parameters. Crystallographic data and selected details of structure determinations are given in Table 1.Acknowledgements
S. S. is indebted to CSIR, India for his Senior Research Fellowship [08/003(0083)/2011-EMR-1]. P. B. acknowledges CSIR-India for the Project (Sanction letter no. 01(2459)/11/EMR-II dated 16/05/2011). Authors are thankful to Prof. Bibhutosh Adhikary, Department of Chemistry, IIEST, Shibpur for his helpful suggestion during preparation of single crystals and determination of structures. The authors also acknowledge the Sophisticated Analytical Instruments Facility at North Eastern Hill University (SAIF-NEHU) for 1H NMR analysis.References
- (a) E. Block, Angew. Chem., Int. Ed. Engl., 1992, 31, 1135 CrossRef; (b) L. Teuber, Sulfur Rep., 1992, 31, 257 Search PubMed; (c) Y. Kanda and T. Fukuyama, J. Am. Chem. Soc., 1993, 115, 8451 CrossRef CAS; (d) T. E. Creighton and P. Rog, Prog. Biophys. Mol. Biol., 1978, 33, 321 Search PubMed; (e) H. F. Gilbert, Methods Enzymol., 1995, 251, 8 CAS; (f) M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614 CrossRef CAS PubMed; (g) B. Heras, M. Kurz, S. R. Shouldice and J. L. Martin, Curr. Opin. Chem. Biol., 2007, 17, 691 CAS; (h) H. Kadokura, F. Katzen and J. Beckwith, Annu. Rev. Biochem., 2003, 72, 111 CrossRef CAS PubMed.
- (a) N. A. Eckardt, Plant Cell, 2006, 18, 1782 CrossRef; (b) Y. Balmer, W. H. Vensel, N. Cai, W. Manieri, P. Schürmann, W. J. Hurkmanand and B. B. Buchanan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2988 CrossRef CAS PubMed.
- (a) R. J. Cremlyn, An Introduction to Organosulfur Chemistry, Wiley-VCH, New York, 1996 Search PubMed; (b) H.-W. Engels, in Ullmann's Encyclopedia of Industrial Chemistry, ed. B. Elvers, S. Hawkins, W. Russey and G. Schulz, Cambridge, New York, 5th edn, 1993, vol. 23, p. 365 Search PubMed; (c) Organic Sulfur Chemistry: Structure and Mechanism, ed. S. Oae, CRS Press, Boca Raton FL, USA, 1991 Search PubMed; (d) G. H. Whitham, Organosulfur chemistry, Oxford University Press, New York, 1995 Search PubMed; (e) D. L. Holbrook, Handbook of Petroleum Refining Processes, ed. R. A. Meyers, McGraw-Hill, New York, 1996, ch. 11.3 Search PubMed; (f) E. Kuhle and E. Klauke, Angew. Chem., Int. Ed. Engl., 1977, 16, 735 CrossRef CAS PubMed.
- J. Srogl, J. Hývl, Á. Révészb and D. Schröder, Chem. Commun., 2009, 3463 RSC.
- (a) A. L. Schwan and R. R. Strickler, Org. Prep. Proced. Int., 1999, 31, 579 CrossRef CAS; (b) W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC; (c) Y. Liu, H. Wang, J. Zhang, J.-P. Wan and C. Wen, RSC Adv., 2014, 4, 19472 RSC.
- (a) A. Shaabani, F. Tavasoli-Rad and D. G. Lee, Synth. Commun., 2005, 35, 571 CrossRef CAS; (b) E. P. Papadopoulos, A. Jarrar and C. H. Issidoides, J. Org. Chem., 1966, 31, 615 CrossRef CAS; (c) A. V. Joshi, S. Bhusare, M. Baidossi, N. Qafisheh and Y. Sasson, Tetrahedron Lett., 2005, 46, 3583 CrossRef CAS; (d) C. Lopez, A. Conzales Cossio and C. Palomo, Synth. Commun., 1985, 15, 1197 CrossRef CAS; (e) S. Raghavan, A. Rajender, S. C. Joseph and M. A. Rasheed, Synth. Commun., 2001, 31, 1477 CrossRef CAS; (f) J. Choi and N. M. Yoon, J. Org. Chem., 1995, 60, 3266 CrossRef CAS; (g) J. B. Arterburm, M. C. Perry, S. L. Nelson, B. R. Dible and M. S. Holguin, J. Am. Chem. Soc., 1997, 119, 9309 CrossRef; (h) K. Tanaka and K. Ajiki, Tetrahedron Lett., 2004, 45, 5677 CrossRef CAS; (i) E. J. Corey and D. L. Boger, Tetrahedron Lett., 1978, 28, 2461 CrossRef; (j) M. N. Bhattacharjee, M. K. Chauduri and H. S. Dasgupta, Synthesis, 1982, 588 CrossRef CAS; (k) A. Christoforou, G. Nicolaou and Y. Elemes, Tetrahedron Lett., 2006, 47, 9211 CrossRef CAS; (l) M. H. Ali and M. McDermott, Tetrahedron Lett., 2002, 43, 6271 CrossRef CAS; (m) P. Zhong and M. P. Guo, Synth. Commun., 2001, 31, 1825 CrossRef CAS; (n) A. Alam, Y. Takaguchi and S. Tsuboi, Synth. Commun., 2005, 35, 1329 CrossRef CAS; (o) D. H. R. Barton, S. V. Ley and C. A. Meerholz, Chem. Commun., 1979, 755 RSC; (p) S. V. Ley, C. A. Meerholz and D. H. R. Barton, Tetrahedron, 1981, 37, 213 CrossRef; (q) D. H. R. Barton, J.-P. Finet and M. Thomas, Tetrahedron, 1986, 42, 2319 CrossRef CAS; (r) N. X. Hu, Y. Aso, T. Otsubo and F. Ogura, Tetrahedron Lett., 1986, 27, 6099 CrossRef CAS.
- (a) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476 RSC; (b) A. Corma, T. Ródenas and M. J. Sabater, Chem. Sci., 2012, 3, 398 RSC; (c) M. Oba, K. Tanaka, K. Nishiyama and W. Ando, J. Org. Chem., 2011, 76, 4173 CrossRef CAS PubMed; (d) P. Das, S. Ray, A. Bhaumik, B. Banerjee and C. Mukhopadhyay, RSC Adv., 2015, 5, 6323 RSC; (e) A. Talla, B. Driessen, N. J. W. Straathof, L.-G. Milroy, L. Brunsveld, V. Hessel and T. Noël, Adv. Synth. Catal., 2015, 357, 2180 CrossRef CAS; (f) S. M. S. Chauhan, A. Kumar and K. A. Srinivas, Chem. Commun., 2003, 2348 RSC; (g) S. Thurow, V. A. Pereira, D. M. Martinez, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Tetrahedron Lett., 2011, 52, 640 CrossRef CAS; (h) K. T. Liu and Y. C. Tong, Synthesis, 1978, 669 CrossRef CAS; (i) N. Iranpoor and B. Zeynizadeh, Synthesis, 1994, 49 Search PubMed; (j) T. Chatterjee and B. C. Ranu, RSC Adv., 2013, 3, 10680 RSC; (k) M. A. Walters, J. Chaparro, T. Siddiqui, F. Williams, C. Ulku and A. L. Rheingold, Inorg. Chim. Acta, 2006, 359, 3996 CrossRef CAS; (l) T. V. Rao, K. N. Rao, S. L. Jain and B. Sain, Synth. Commun., 2002, 32, 1151 CrossRef CAS; (m) L. I. Simandi, S. Nemeth and N. Rumelis, J. Mol. Catal., 1987, 42, 357 CrossRef CAS; (n) A. Cervilla, A. Corma, V. Fornés, E. Llopis, P. Palanca, F. Reyy and A. Ribera, J. Am. Chem. Soc., 1994, 116, 1595 CrossRef CAS; (o) A. Saxena, A. Kumary and S. Mozumdar, J. Mol. Catal. A: Chem., 2007, 269, 35 CrossRef CAS.
- (a) H. T. Abdel-Mohsen, K. Sudheendran, J. Conrad and U. Beifuss, Green Chem., 2013, 15, 1490 RSC; (b) J. L. G. Ruano, A. Parra and J. Alemán, Green Chem., 2008, 10, 706 RSC; (c) Y. Liu, H. Wang, C. Wang, J.-P. Wan and C. Wen, RSC Adv., 2013, 3, 21369 RSC; (d) H. Wang, G. Huang, Y. Sun and Y. Liu, J. Chem. Res., 2014, 38, 96 CrossRef CAS.
- (a) A. R. Katritzky, Handbook of Heterocyclic Chemistry, Pergamon Press, Oxford, 1986 Search PubMed; (b) N. Saracoglu, Tetrahedron, 2007, 63, 4199 CrossRef CAS; (c) G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299 CrossRef CAS PubMed; (d) L. Fritea, P. Audebert, L. Galmiche, K. Gorgy, A. Le Goff, R. Villalonga, R. Săndulescu and S. Cosnier, ChemPhysChem, 2015, 16, 3695 CrossRef CAS PubMed.
- W. Kaim, Coord. Chem. Rev., 2002, 230, 127 CrossRef CAS.
- (a) B. J. Jordan, M. A. Pollier, L. A. Miller, C. Tiernan, G. Clavier, P. Audebert and V. M. Rotello, Org. Lett., 2007, 9, 2835 CrossRef CAS PubMed; (b) Y.-H. Gong, P. Audebert, J. Tang, F. Miomandre, G. Clavier, S. Badré, R. Méallet-Renault and J. Marrot, J. Electroanal. Chem., 2006, 592, 147 CrossRef CAS.
- S. Fukuzumi, J. Yuasa and T. Suenobu, J. Am. Chem. Soc., 2002, 124, 12566 CrossRef CAS PubMed.
- (a) S. Samanta, S. Das and P. Biswas, J. Org. Chem., 2013, 78, 11184 CrossRef CAS PubMed; (b) S. Samanta and P. Biswas, RSC Adv., 2015, 5, 84328 RSC.
- C. A. Beach, D. C. Mays, R. C. Guiler, C. H. Jacober and N. Gerber, Clin. Pharmacol. Ther., 1986, 39, 265 CrossRef CAS PubMed.
- (a) G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299 CrossRef CAS PubMed; (b) P. Audebert, S. Sadki, F. Miomandre, G. Clavier, M. C. Vernières, M. Saoud and P. Hapiot, New J. Chem., 2004, 28, 387 RSC; (c) M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263 CrossRef CAS PubMed.
- R. Hosseinzadeh, M. Tajbakhsh, H. Khaledi and K. Ghodrati, Monatsh. Chem., 2007, 138, 871 CrossRef CAS.
- E. Rattanangkool, W. Krailat, T. Vilaivan, P. Phuwapraisirisan, M. Sukwattanasinitt and S. Wacharasindhu, Eur. J. Org. Chem., 2014, 4795 CrossRef CAS.
- R. S. Varma, H. M. Meshramzt and R. Dahiyal, Synth. Commun., 2000, 30, 1249 CrossRef CAS.
- SAINT, version 6.02; SADABS, version 2.03, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed.
- SHELXTL, version 6.10, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1, 1H and 13C NMR spectra, CIFs. CCDC 1437412 and 1437413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra01509c |
| This journal is © The Royal Society of Chemistry 2016 |