
Hydrogels from amphiphilic star block copolypeptides†
R. Murphyab,
T. Borasea,
C. Paynec,
J. O'Dwyerc,
S.-A. Cryancde and
A. Heise*ab
aPolymer Chemistry and Biopolymers Laboratory, Department of Pharmaceutical and Medicinal Chemistry, Royal College of Surgeons in Ireland, Dublin 2, Ireland. E-mail: andreasheise@rcsi.ie
bSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland
cSchool of Pharmacy, Royal College of Surgeons in Ireland, Dublin 2, Ireland
dTissue Engineering Research Group, Royal College of Surgeons in Ireland, Ireland
eTrinity Centre for Bioengineering, Trinity College Dublin, Dublin 2, Ireland
First published on 23rd February 2016
Abstract
A range of 8-arm star-shaped diblock copolypeptides were developed to investigate their potential as self-assembling amphiphiles in aqueous media. These star-shaped block copolypeptides were sythesised by sequential N-carboxyanhydride (NCA) polymerisation from a polypropylene imine (PPI) dendrimer core to comprise a poly(L-lysine) core and poly(L-tyrosine), poly(L-phenylalanine), poly(L-valine) and poly(L-alanine) outer blocks. All star block copolypeptides except the poly(alanine) copolymer spontaneously self-assemble into hydrogels at a low polymer concentration of ≤2 wt%. Investigation of the secondary structure by FTIR and Circular Dichroism (CD) spectroscopy confirm a largely random coil conformation. The hydrogels were further investigated for their viscoelastic properties revealing a strong structure dependence of the hydrogel strength. The highest gel strength was achieved with the star block copolypeptide containing the poly(L-phenylalanine) outer blocks. The versatility of the hydrophilic/hydrophobic system allows for straightforward modification of the hydrophobic sequence, which can be utilised to modulate the mechanical strength for various applications.
1 Introduction
Hydrogels are highly versatile materials in biomedical science. Based on their high water content and permeability as well as their structural integrity similar to the extracellular matrix, hydrogels are ideal building blocks in regenerative medicine and as therapeutic delivery systems.1–4 Continued development of more efficient synthetic methodologies has enabled the design of a class of potentially biocompatible hydrogelators in the form of structurally well-defined oligopeptide amphiphiles.5 Supramolecular self-assembly of oligopeptide amphiphiles into interconnected hydrogel networks is known to be stabilised via hydrogen bonding and other non-covalent interactions such as electrostatic interactions, π–π stacking and hydrophobic interactions.6–9Polypeptide structures readily obtained by the polymerization of amino acid N-carboxyanhydrides (NCA) have also been explored for hydrogel formation. For example, Deming presented the design of hydrogels from linear amphiphilic co-polypeptides.10–13 One such report details the hierarchical self-assembly of these materials composed of a hydrophobic poly(L-leucine) segment grafted to a cationic poly(L-lysine) block. Self-assembly was attributed to the specific peptide secondary conformation, whereby adjacent lysine sequences form long fibrous networks while poly(L-leucine) residues adopt a rigid α-helical arrangement driving spontaneous hydrogel formation in an aqueous environment. Subsequent in vivo studies have demonstrated the suitability of these block copolypeptides as potential scaffolds or delivery matrixes in CNS tissue.14 We recently disclosed an NCA-derived PEG2000-Tyr5 amphiphilic block copolymer that can undergo thermoresponsive hydrogelation at a low concentration range of 0.25–3.0 wt% within a temperature range of 25 to 50 °C.15 While this material displays a desirable inverse thermoresponse (i.e. it hydrogelates upon temperature increase), it is extremely sensitive to structural variation, which limits the scalability and applicability of the material. We hypothesize that a pre-organisation of the polypeptide amphiphiles might overcome these disadvantages while maintaining a low solid content in the hydrogel. Pre-organisation was first utilized by Jeong who reported a series of amphiphilic hybrid PEG block copolymers obtained by ring-opening polymerization of alanine or phenylalanine NCAs.16–21 These materials self-assemble into nanostructures such as micelles in aqueous solutions and further transition into hydrogels at high polymer concentration in the range of 3.0–14 wt% through intermicellar aggregation when the temperature increases. However, the initial step of micelle formation is a dynamic process and strongly dependents on the conditions and concentration of the amphiphiles.
Here we propose amphiphilic star polypeptides as hydrogelators in which the individual amphiphiles are “fixed” to a central core thus providing a covalent core–shell nanoparticle similar to micelles. Star polypeptide synthesis by NCA polymerisation has significantly advanced recently22–25 and several groups have evaluated these higher order polypeptide structures for the delivery of therapeutics.26–28 Only few examples of star polypeptide hydrogels have been reported thus far. Li prepared a series of poly(L-diethylene glycol glutamate) (L-EG2Glu) sequences grafted from 8-arm and 32-arm dendritic molecules.29 Highly dependent on arm and sequence composition, the star polymers underwent transformation into hydrogel networks at low critical gelation concentration (CGC) ranging from 1–3 wt%. The lower CGC threshold could only be observed using the 32-arm polypeptides, which recorded a notably higher polydispersity index (PDI) than the 8-arm analogues. Thornton has also reported the generation of polypeptide hydrogels comprising of a star-shaped architecture.30 Synthesised via the polymerisation of alanine NCA from a 4-armed star-shaped poly(ethylene glycol) (PEG) initiator, the hydrogel could form at about 10 wt% and was found to be enzymatically degradable.
Herein we present the synthesis of a series of novel star-shaped diblock copolypeptide amphiphiles, affording materials which are spontaneous forming hydrogels. The use of the highly branched star architectures allows for superior intermolecular physical interactions between the adjacent hydrophobic moieties when compared with their linear counterparts. Close examination of the conformation via spectroscopic analysis revealed that the star macromolecular architecture appears to be governed by the influence of a range of hydrophobic residues, i.e. methyl (L-alanine), isopropyl (L-valine), phenyl (L-phenylalanine) and phenol (L-tyrosine) groups. The disparate chemistry attached to the individual moieties promotes contrasting self-assembling properties in the series of star polypeptide amphiphiles. This enables a seamless modification of hydrogel strength by alternating the hydrophobic side chain.
2 Experimental
Materials
All chemical were obtained from Sigma-Aldrich unless other wise noted. ε-Carbobenzyloxy-L-lysine, L-phenylalanine, O-benzyl-L-tyrosine, L-alanine and L-valine were purchased from Bachem. Second generation (G2) poly-propylene imine (PPI) dendrimer was obtained from SyMO-Chem BV (The Netherlands). The NCAs of ε-carbobenzyloxy-L-lysine, L-phenylalanine, O-benzyl-L-tyrosine, L-alanine and L-valine were synthesised following literature procedures.15,32Methods
Nuclear Magnetic Resonance (NMR) analysis was completed using a Bruker Avance 400 (400 MHz) spectrometer at room temperature with d-DMSO, D2O, CDCl3 and d-TFA as solvents. Attenuated total reflection (ATR) FT-IR spectra were recorded using a Perkin-Elmer Spectrum 100 in the region of 4000–650 cm−1. Four scans were completed with a resolution of 2 cm−1. A background measurement was performed prior to loading the sample onto the ATR for measurement. Size Exclusion Chromatography (SEC) was performed on an Agilent 1200 system in combination with two PSS GRAM analytical (8 × 300 and 8 × 100, 10 μ) columns, a Wyatt Dawn Heleos 8 multi angle light scattering detector (MALS) and a Wyatt Optilab rEX differential refractive index detector (DRI) with a 658 nm light source. The eluent was DMF containing 0.1 M LiBr at a flow rate of 1 mL min−1. The column temperature was set to 40 °C with the MALS detector at 35 °C and the DRI detector at 40 °C. Molar masses and polydispersities were calculated from the MALS signal by the Astra software (Wyatt) using the refractive index increment (dn/dc) of 0.101 for ply-ε-carbobenzyloxy-L-lysine (PZLL). All GPC samples were prepared using a concentration of 2 mg mL−1 and were filtered through a 0.45 μm millipore filter, prior to injection. Circular dichroism (CD) spectra were recorded on an Aviv CD Spectrophotometer 410 (Aviv Biomedical, Inc. New Jersey, USA). The star copolypeptides were prepared at a concentration of 0.3 mg mL−1 in deionised water. Each solution was placed into a quartz cell with a path length of 0.1 cm and analysed in the UV region (180 to 280 nm). Deionised water was used as a reference for baseline correction before sample measurement. The mean residue ellipticity was calculated using the following equation:| [Θ]MRW/(10 × l × c) | (1) |
With experimentally determined (Θ) in mdeg, mean residue weight (MRW) in g mol−1, path length (l) in cm and sample concentration (c) in mg mL−1. The existence of α-helical conformation can be calculated from the following equation:
% α-helix = (−[Θ222] + 3000)/39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000
| (2) |
| Q = (ms − mr)/mr | (3) |
Star-shaped poly(ε-carbobenzyloxy-L-lysine-b-benzyl-L-tyrosine) (PLL40-b-PLT5, representative procedure)
The NCA of ε-carbobenzyloxy-L-lysine (2.37 g, 7.74 mmol) was added to a Schlenk flask. Under a N2 atmosphere, 35 mL of anhydrous CHCl3 was added until the NCA was dissolved. The flask was placed in a temperature controlled oil/water bath at 0 °C. G2 PPI dendrimer (18 mg, 2.42 × 10−2 mmol) was prepared in 2 mL of dry CHCl3, and quickly added to the Schlenk flask via syringe. The solution was allowed to stir for 24 hours at 0 °C. FT-IR analysis was utilised to confirm total consumption of NCA monomer. A sample was taken directly via syringe to monitor the molecular mass using GPC. The NCA of O-benzyl-L-tyrosine (288 mg, 9.68 × 10−1 mmol) was dissolved in 5 mL anhydrous CHCl3 and charged to the Schlenk flask via syringe. The solution was allowed to stir at 0 °C until all of the NCA monomer was consumed as confirmed by FT-IR. The polymer was precipitated into excess diethyl ether and dried under vacuum in a dessicator (yield 89%). Other star block copolymers were prepared accordingly replacing the O-benzyl-L-tyrosine NCA with L-phenylalanine NCA, L-alanine NCA and L-valine NCA.Deprotection of star-shaped block copolymers (representative procedure for PLL40-b-PLT5)
1 g of star PLL40-b-PLT5 was dissolved in 11 mL of trifluoro acetic acid (TFA) and allowed to stir for 1 h. 5 mL of HBr 33% wt in acetic acid was added drop wise to the solution in a six-fold excess with respect to ε-carbobenzyloxy-L-lysine and O-benzyl-L-tyrosine and allowed to stir for 15 h. The polymer was precipitated twice into diethyl ether (40 mL) and centrifuged. The TFA/diethyl ether waste was decanted and the precipitate was washed once more with diethyl ether. The polymer was dried under vacuum and then subsequently dissolved in deionised water. Dialysis was performed against deionised water for 4 days using a 3500 MWCO membrane, with frequent water replacement. The polymer was then lyophilised (yield 65%). Deprotection of the other block copolymers was carried out with a reduced quantity of HBr as PZLL was the only sequence present bearing cleavable protecting groups.3 Results and discussion
Synthesis and hydrogelation
The general design of the 8-arm star polypeptide hydrogelators comprises a core region of hydrophilic poly(L-lysine) (PLL) blocks extended with hydrophobic oligopeptide blocks. The latter were selected from a range of amino acids capable of exhibiting differed modes of interaction and hydrophobicities to elucidate the structural effect on the hydrogel formation. Elaborating on previous work from our group,24 an 8-arm primary amine terminated PPI dendrimer was used as an initiator in the ring opening polymerization of ε-carbobenzyloxy-L-lysine (ZLL or cbz-L-lysine) NCA at a constant PPI to ZLL-NCA ratio of 1:320 (i.e. targeting 40 units per arm) using high vacuum techniques at 0 °C.32 Subsequent macroinitiation of hydrophobic amino acid NCAs of benzyl-L-tyrosine (BLT), L-phenyl alanine (LPA), L-alanine (LA) and L-valine (LV) was accomplished after full ZLL NCA conversion (confirmed by FTIR) without intermediate work-up targeting an average block length of 5 units per arm (Scheme 1, Table 1). The block length ratio of 40:5 was identified as the ideal balance between hydrophilic and hydrophobic blocks in initial gelling experiments. The choice of amino acids is rationalized by the fact that the poly(LPA) block bearing phenyl functionalities in comparison to the poly(LT)'s phenol moieties. Moreover, both poly(LA) and poly(LV) peripheral sequences bear aliphatic methyl and isopropyl groups, respectively, without the possibility of aromatic π–π interaction.
 | ||
| Scheme 1 | ||
| Polymera | Mn (g mol−1) theor. | Mn (g mol−1) SEC | PDI |
|---|---|---|---|
| a Indices denote the theoretical composition (NCA/R–NH2 ratio).b The indices for this sample denote the degree of polymerisation from SEC analysis as full monomer conversion was not achieved. | |||
| 8-PZLL40 | 83800 |
82300 |
1.04 |
| 8-PZLL40-b-PBLT5 | 94000 |
93400 |
1.07 |
| 8-PZLL40 | 83800 |
70900 |
1.05 |
| 8-PZLL40-b-PLPA5 | 89600 |
79400 |
1.09 |
| 8-PZLL40 | 83800 |
67900 |
1.03 |
| 8-PZLL40-b-PLV5 | 87900 |
77500 |
1.05 |
| 8-PZLL40 | 83800 |
60600 |
1.03 |
| 8-PZLL40-b-PLA5 | 86600 |
63600 |
1.04 |
| L-PZLL32 | 10400 |
8300 | 1.04 |
| 1-PZLL32-b-PBLT5 | 11200 |
9500 | 1.11 |
| L-PZLL148b | 83800 |
38700 |
1.02 |
| 1-PZLL148-b-PBLT30 | 94000 |
46600 |
1.20 |
Successful grafting of PZLL from the PPI core was confirmed by 1H-NMR spectroscopy (ESI Fig. S1 and S2†). After addition of the second block, size excluison chromatography (SEC) confirmed the anticipated increase in molecular weight for all star diblock copolypeptides (Fig. 1 and Table 1; ESI Fig. S4–S6†).
 | ||
| Fig. 1 SEC traces of 8-PZLL40-b-PLPA5 (A), 8-PZLL40 (B); 8-PZLL40-b-PLV5 (C) and 8-PZLL40 (D). | ||
Quantitative deprotection of the PZLL inner block was achieved with TFA and HBr 33% wt in acetic acid, which in the case of PBLT simultaneously deprotected the tyrosine benzyl ether evident from the quantitative disappearance of the aromatic peak at 7.2 ppm in the 1H-NMR spectra (ESI Fig. S3†) yielding amphiphilic star block copolymers 8-PZLL40-b-PBLT5, 8-PLL40-b-PLT5, 8-PLL40-b-PLV5 and 8-PLL40-b-PLPA5. Differences in hydrogelation capacity became immediately apparent. Deprotected 8-arm PLL (8-PLL40) alone did not form hydrogels in water, while the tyrosine extended star block copolymer 8-PLL40-b-PLT5 formed transparent hydrogels at concentrations as low as 2.0 wt%. Both 8-PLL40-b-PLPA5 and 8-PLL40-b-PLV5 also spontaneously formed hydrogels in contrast to 8-PLL40-b-PLA5, which did not form hydrogels at any concentration (Fig. 2). Upon observation, 8-PLL40-b-PLT5 was visibly more transparent than the other samples, which appear slightly opaque. In an attempt to investigate the influence of the star-shaped architecture on the hydrogel formation the synthesis of a linear analogue L-PLL320-b-PLT40 with exactly the same overall molecular weight and L-PLL40-b-PLT5 representative of a single arm was attempted. While the latter could be synthesized, limitations of the amine initiation in realizing high molecular weights only permitted the synthesis of a linear equivalent L-PZLL148-b-PBLT30. Notably, neither linear analogue underwent hydrogelation highlighting the importance of the star architecture in this process.
 | ||
| Fig. 2 Hydrogels obtained from 8-PLL40-b-PLT5 (A), 8-PLL40-b-PLV5 (B) and 8-PLL40-b-PLPA5 (C) at a polymer concentration of 3 wt%. | ||
In order to elucidate the morphology of the star diblock copolypeptide hydrogels, samples were freeze dried in the gelled state and imaged using Scanning Electron Microscopy (SEM) (Fig. 3). Condensed fibers are observed in lyophilized 8-PLL40-b-PLT5, revealing a highly porous structure with fibers ∼25.7 μm apart. While 8-PLL40-b-PLPA5 exhibits a ribbon-like network with twisting layers developing into a small porous structure characteristic of β-sheet conformation.33 Observed in 8-PLL40-b-PLV5 is an elongated network with pore sizes as large as 37.3 μm and supermolecular fibers stacking along a single axis.
 | ||
| Fig. 3 SEM images of lyophilised hydrogels obtained from 8-PLL40-b-PLT5 ((A), pore size 25.7 μm), 8-PLL40-b-PLV5 ((B), pore size 37.3 μm) and 8-PLL40-b-PLPA5 ((C), pore size 5.9 μm) at a polymer concentration of 3 wt%. Scale bars are 100 μm. | ||
Secondary structure analysis
The conformational analysis of each star diblock polypeptide was done using ATR-FTIR in the dry state after lyophilisation and Circular Dichroism (CD) in the gel state. In the dry state all star polypeptides display an intense amide I band around 1647 cm−1, characteristic of a dominant random coil conformation. In addition, small differences in the band structure around the amide I and amid II bands are apparent, which suggests the coexistence of β-motifs (ESI Fig. S7†). For example, in the 8-PLL40-b-PLT5 spectrum an additional amide I shoulder peak is visible at 1677 cm−1, suggestive of β-turn. The other gel forming polypeptides 8-PLL40-b-PLPA5 and 8-PLL40-b-PLT5 also exhibited a split amide I band (1628 cm−1 and 1648 cm−1). The 1648 cm−1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration can be assigned to random coil structures, while the presence of β-sheets is suggested by the band at 1628 cm−1. Also in this case a shoulder peak at 1674 cm−1 could be assigned to the presence of β-turn conformation. The co-occurrence of random coil, β-sheet and β-turn configurations seems conceivable considering the complex star shaped polypeptide architecture. Hierarchal stacking of the hydrophobic moieties can occur through intermolecular and intramolecular interactions. Evident β-sheet structures here can be attributed to intermolecular stacking of residues akin to the parallel or twisting patterns.34,35 The β-turn motif is plausible since oligopeptide sequences may fold ‘back on themselves’ through hydrogen bonding 3–4 residues apart.36,37 Both stacking configurations are comparable to patterns observed in micelle formation considering the architecture of the star polypeptide.38–40 Notably, there is an absence of β-sheet bands in non-hydrogelling 8-PLL40-b-PLA5. Observed are vibrational modes located at 1649 cm−1 and 1543 cm−1, representative of a random coil population. In the absence of β-sheet structures no intramolecular network can be formed. The importance of the coexistence of two conformations for hydrogel formation was highlighted in the literature albeit for linear PLL block copolymers adopting β-sheets and α-helices.31
O vibration can be assigned to random coil structures, while the presence of β-sheets is suggested by the band at 1628 cm−1. Also in this case a shoulder peak at 1674 cm−1 could be assigned to the presence of β-turn conformation. The co-occurrence of random coil, β-sheet and β-turn configurations seems conceivable considering the complex star shaped polypeptide architecture. Hierarchal stacking of the hydrophobic moieties can occur through intermolecular and intramolecular interactions. Evident β-sheet structures here can be attributed to intermolecular stacking of residues akin to the parallel or twisting patterns.34,35 The β-turn motif is plausible since oligopeptide sequences may fold ‘back on themselves’ through hydrogen bonding 3–4 residues apart.36,37 Both stacking configurations are comparable to patterns observed in micelle formation considering the architecture of the star polypeptide.38–40 Notably, there is an absence of β-sheet bands in non-hydrogelling 8-PLL40-b-PLA5. Observed are vibrational modes located at 1649 cm−1 and 1543 cm−1, representative of a random coil population. In the absence of β-sheet structures no intramolecular network can be formed. The importance of the coexistence of two conformations for hydrogel formation was highlighted in the literature albeit for linear PLL block copolymers adopting β-sheets and α-helices.31
CD spectroscopy was utilised to gather information about the asymmetric arrangement of the peptide backbone in the hydrogel state. The CD spectra of all polypeptides are collated in Fig. 4. All of the star architectures exhibited a broad negative trough below 200 nm and an intense positive peak above 215 nm in agreement with the FTIR results of a random coil configuration. The highest random coil content was observed for 8-PLL40-b-PLV5 and 8-PLL40-b-PLPA5. The decreasing band intensity of the 8-PLL40-b-PLT5 minima at 195 nm suggest that the random coil content is affected by the hydroxyl substituent on the phenyl ring. The random coil contribution is further reduced in the non-gelling 8-PLL40-b-PLA5 as well as in the linear reference polypeptide. Notably, the degree of unordered conformation in 8-PLL40-b-PLV5 is sufficient enough to promote hydrogelation. However, the same conformational abundance is deficient in 8-PLL40-b-PLA5, explaining the absence of self-assembly. The CD spectra are devoid of any characteristic β-sheet or β-turn peaks, which would be evident by maxima at ∼200 nm and ∼220 nm and minima at ∼206 nm.41 The common feature of coil conformation observed from the spectroscopic analysis indicates that PLL regulates the folding pattern due to its excess with respect to the hydrophobic sequence.
 | ||
| Fig. 4 Circular dichroism (CD) spectra of amphiphilic star block copolymers in water. Assignment of spectra is indicated from bottom to top of the minima. | ||
Hydrogel properties
The extent of gelation of the star-shaped polypeptides was determined from comparison of dry and swollen samples. The minimum gelation concentration (MGC) was determined from incremental increase in water concentration which was followed by time equilibrated gelation (Table 2). The transition from sol to gel for 8-PLL40-b-PLT5 was noted at 2.0 wt% polymer content using the vial inversion method. In contrast 8-PLL40-b-PLPA5 still maintained its crosslinked network at a higher water concentration (98.75%). A low MGC was also observed in the 8-PLL40-b-PLV5 sample (1.5 wt%). The degree of swelling of the star block copolypeptides measured with a solid content of 3.0 wt% did not reveal significant differences in the equilibrium weight swelling ratios (∼50–55%; Table 2).| Polypeptide | EGTa (s) | MGC (wt%) | Qb | G′ (Pa) | Injection force (N) |
|---|---|---|---|---|---|
| a Time from initial dispersion in aqueous media until complete dissolution with gelation confirmed using the vial inversion method.b 3.0 wt% in deionised water; Q = (ms − mr)/mr, where mr is the hydrogel mass in dry state and ms is the hydrogel mass in the swollen state. | |||||
| 8-PLL40-b-PLT5 | 1200 | 2.00 | 50.76 | 180.7 | 5.88 |
| 8-PLL40-b-PLPA5 | 600 | 1.25 | 55.69 | 2210.1 | 7.29 |
| 8-PLL40-b-PLV5 | 350 | 1.50 | 49.88 | 220.5 | 7.22 |
An interesting feature of these materials is their stability in salt solution. For reference, only Deming has previously demonstrated the stability of cationic polypeptide hydrogels in both high and low ionic strength media.11 Although alternative secondary structural configuration contributes to self-assembly here, the hydrogels exhibit similar properties over a small range of ionic concentrations (Fig. S8†). The swollen gel networks appear unhindered over this range, although a rheological assay may provide a better insight into the true ‘masking effect’ of NaCl on the PLL electrolyte segments. A reduction in the degree of swelling is prompted after incubation in saturated 10−2 M NaCl solution and was noted in accordance with increasing visual turbidity. Thus, the deterioration of the gel network of all star polypeptide samples became apparent. Media began to separate from the gel and collapse of the network ensued after equilibrium incubation.
The rheological properties of all star block copolypeptides hydrogels were measured at the same polymer concentration (3.0 wt%) in water in order to determine the viscoelasticity of the samples. Initially, the linear viscoelastic regime was assessed by a strain sweep experiment (Fig. 5B). A frequency sweep experiment provided insight into both the storage (G′) and loss modulus (G′′) of the material (Fig. 5A). The 8-PLL40-b-PLT5 hydrogel exhibited a storage modulus of 168 Pa, which was found to be the weakest polypeptide hydrogel. Altering the chemistry of the side group by removing the hydroxyl group had a significant effect on the mechanical properties. To illustrate the sizeable gap in gel strength, the storage modulus increased to 2040 Pa for 8-PLL40-b-PLPA5 hydrogel. The difference of ∼1870 Pa between its L-tyrosine analogue illustrates the highly sensitive nature of physical crosslinking sites within these star-shaped hydrogel networks.
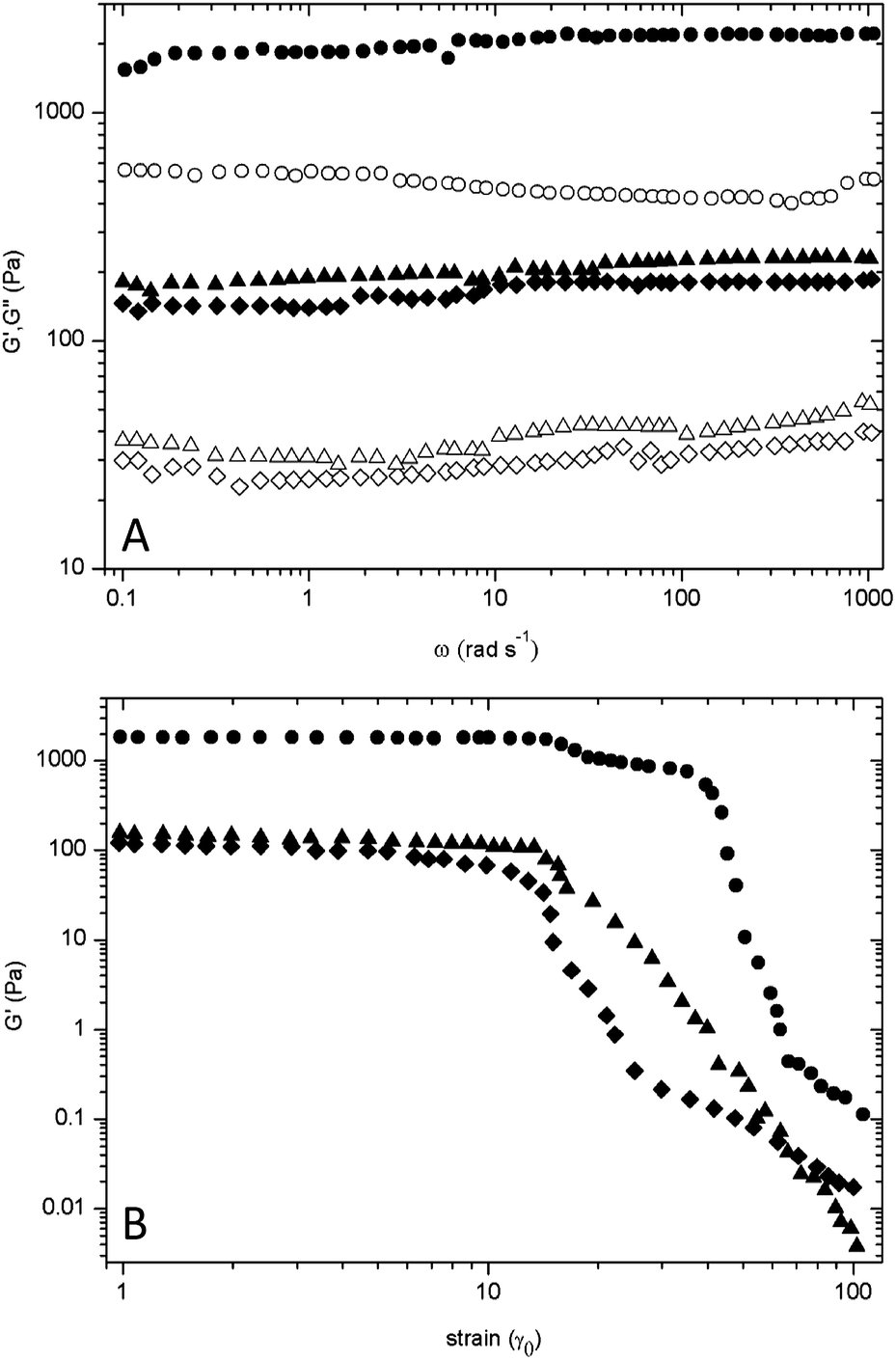 | ||
| Fig. 5 Rheological properties of star diblock copolypeptides, 8-PLL40-b-PLPA5 (●), 8-PLL40-b-PLV5 (▲), 8-PLL40-b-PLT5 (◆). (A) Gel strength over linear frequency (sheared at γ = 0.1, ω = 1–1000 rad s−1); closed symbols – storage modulus; open symbols – loss modulus. (B) Gel strength over increasing strain (sheared at γ = 1–100, ω = 1 rad s−1). | ||
It could be hypothesised that incorporation of the hydroxyl group provides a partially solubilising functionality via the acidic proton, which would allow water to penetrate. Upon incremental increase in the strain amplitude (γ = 1–100), shear thinning of each of the hydrogels samples became apparent (Fig. 5B). However, all star diblock copolypeptide hydrogels demonstrated rapid recovery properties after application of a large strain (ESI, Fig. S9†). Initially, the gel structure was broken down under large oscillatory amplitude (γ = 1). The storage modulus G′ of both 8-PLL40-b-PLT5 and 8-PLL40-b-PLV5 underwent a reduction just over one order of magnitude while the G′ of 8-PLL40-b-PLPA5 dropped by two orders of magnitude. After a return to a lower amplitude strain (γ = 0.1) all polypeptide hydrogels recovered to their original gel strength within 5–20 seconds. In particular, 8-PLL40-b-PLPA5 underwent network re-assembly almost instantaneously. These properties validate the hydrogels as suitable candidates for applications through a syringe/needle. Indeed, when the gels were loaded into a syringe only a moderate force of 5.9–7.3 N was required to push them though the attached needle and subsequently recover their gel state (Table 2, Fig. S10 and S11†). The force applied is acceptable when considering compression forces up to 10 N were required for therapeutic pramlintide injections, albeit with slightly smaller needles (29G and 31G).42 The data implies an advantageous feature of these materials, which is that hydrogel strength can be easily modulated for application purposes. The comparative storage and loss moduli of both 8-PLL40-b-PLT5 and 8-PLL40-b-PLV5 hydrogels would be suitable for a drug delivery matrix.43 The high strength associated with the 8-PLL40-b-PLPA5 hydrogel (3 wt% – 2.2 kPa) regards it compatible in applications ranging from wound healing scaffolds to vascular tissue scaffolds.44,45 The polymer 8-PLL40-b-PLPA5 demonstrated superiority in physical crosslinking due to the densely packed aromatic residues. It can be concluded that the inclusion of a hydroxyl substituent on the aromatic ring or the replacement with an aliphatic residue proved to be detrimental to hydrogel strength as the storage modulus decreased almost tenfold in each case.
4 Conclusions
A series of star-shaped amphiphilic diblock copolypeptides with a poly(L-lysine) core were prepared from the ring opening polymerisation of NCA amino acid monomers grafted to an 8-arm PPI dendrimer. Spontaneous aqueous self-assembly was observed for copolymers composed of L-tyrosine, L-phenylalanine and L-valine outer blocks but not with L-alanine blocks or linear model polymers. Sol–gel transitions were observed at particularly lower polymer concentration (1.25 wt%) for star copolymers containing the phenyl functionality. This moiety also appears to contribute to a stronger gel network as seen in the rheological measurements. The unique self-assembly associated with amphiphilic star polypeptides afforded hydrogel materials with multiple properties. They maintain structure with high water content, can be injected with minimal force and are potentially enzymatically degradable, which can be utilised for various applications.Acknowledgements
Financial support from the Science Foundation Ireland (SFI) Principle Investigator Award 13/IA/1840 is gratefully acknowledged.Notes and references
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- A. Altunbas and D. J. Pochan, Top. Curr. Chem., 2012, 310, 135–167 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26 RSC.
- F. J. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160–173 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld, D. J. Pine and T. J. Deming, J. Am. Chem. Soc., 2003, 125, 15666–15670 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld and T. J. Deming, Supramol. Chem., 2006, 18, 423–427 CrossRef CAS.
- Z. Li and T. J. Deming, Soft Matter, 2010, 6, 2546–2551 RSC.
- C. Y. Yang, B. Song, Y. Ao, A. P. Nowak, R. B. Abelowitz, R. A. Korsak, L. A. Havton, T. J. Deming and M. V. Sofroniew, Biomaterials, 2009, 30, 2881–2898 CrossRef CAS PubMed.
- J. Huang, C. L. Hastings, G. P. Duffy, H. M. Kelly, J. Raeburn, D. J. Adams and A. Heise, Biomacromolecules, 2013, 14, 200–206 CrossRef CAS PubMed.
- Y. Y. Choi, J. H. Jang, M. H. Park, B. G. Choi, B. Chi and B. Jeong, J. Mater. Chem., 2010, 20, 3416–3421 RSC.
- H. J. Oh, M. K. Joo, Y. S. Sohn and B. Jeong, Macromolecules, 2008, 41, 8204–8209 CrossRef CAS.
- Y. Jeong, M. K. Joo, K. H. Bahk, Y. Y. Choi, H. T. Kim, W. K. Kim, H. J. Lee, Y. S. Sohn and B. Jeong, J. Controlled Release, 2009, 137, 25–30 CrossRef CAS PubMed.
- E. Y. Kang, B. Yeon, H. J. Moon and B. Jeong, Macromolecules, 2012, 45, 2007–2013 CrossRef CAS.
- U. P. Shinde, M. K. Joo, H. J. Moon and B. Jeong, J. Mater. Chem., 2012, 22, 6072–6079 RSC.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2012, 45, 423–433 Search PubMed.
- A. Sulistio, A. Blencowe, A. Widjaya, X. Zhang and G. Qiao, Polym. Chem., 2012, 3, 224–234 RSC.
- S. Junnila, N. Houbenov, S. Hanski, H. Iatrou, A. Hirao, N. Hadjichristidis and O. Ikkala, Macromolecules, 2010, 43, 9071–9076 CrossRef CAS.
- M. Byrne, D. Victory, A. Hibbitts, M. Lanigan, A. Heise and S. A. Cryan, Biomater. Sci., 2013, 1, 1223–1234 RSC.
- S. Abraham, C. S. Ha and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2774–2783 CrossRef CAS.
- K. Wang, H. Q. Dong, H. Y. Wen, M. Xu, C. Li, Y. Y. Li, H. N. Jones, D. L. Shi and X. Z. Zhang, Macromol. Biosci., 2011, 11, 65–71 CrossRef CAS PubMed.
- M. Byrne, P. D. Thornton, S. A. Cryan and A. Heise, Polym. Chem., 2012, 3, 2825–2831 RSC.
- H. M. Wu, S. R. Pan, M. W. Chen, Y. Wub, C. Wang, Y. T. Wen, X. Zeng and C. B. Wu, Biomaterials, 2011, 32, 1619–1634 CrossRef CAS PubMed.
- Y. Shen, S. Zhang, Y. Wan, W. Fu and Z. Li, Soft Matter, 2015, 15, 2945–2951 RSC.
- P. D. Thornton, S. M. Reduwan Billah and N. R. Cameron, Macromol. Rapid Commun., 2013, 34, 257–262 CrossRef CAS PubMed.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- G. J. Habraken, M. Peeters, C. H. Dietz, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 514–524 RSC.
- M. A. Poirier, H. Jiang and C. A. Ross, Hum. Mol. Genet., 2005, 14, 765–774 CrossRef CAS PubMed.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS PubMed.
- V. Moretto, M. Crisma, G. M. Bonora and C. Toniolo, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- K. Rajagopal, B. Ozbas, D. J. Pochan and J. P. Schneider, Eur. Biophys. J., 2006, 35, 162–169 CrossRef CAS PubMed.
- E. G. Hutchinson and J. M. Thornton, Protein Sci., 1994, 3, 2207–2216 CrossRef CAS PubMed.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- M. W. Werten, A. P. Moers, T. Vong, H. Zuilhof, J. C. van Hest and F. A. de Wolff, Biomacromolecules, 2008, 9, 1705–1711 CrossRef CAS PubMed.
- D. Merritt, B. Schreiner, S. Harris, M. B. DeYoung, S. Strobel and J. Lauinger, J. Diabetes Sci. Technol., 2010, 4, 1438–1446 CrossRef PubMed.
- F. Rehfeldt, A. J. Engler, A. Eckhardt, F. Ahmed and D. E. Discher, Adv. Drug Delivery Rev., 2007, 59, 1329–1339 CrossRef CAS PubMed.
- A. Song, A. A. Rane and K. L. Christman, Biomaterials, 2007, 28, 567–575 CrossRef PubMed.
- A. C. Jiminez-Vergara, V. Guiza-Arguello, S. Becerra-Bayona, D. J. Munoz-Pinto, R. E. McMahon, A. Morales, L. Cubero-Ponce and M. S. Hahn, Ann. Biomed. Eng., 2010, 38, 2885–2895 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEC traces, FTIR spectra, 1H-NMR spectra, additional rheology plots. See DOI: 10.1039/c6ra01190j |
| This journal is © The Royal Society of Chemistry 2016 |