
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical synthesis of a nanohybrid film consisting of stacked graphene sheets and manganese oxide as oxygen evolution reaction catalyst†
Masaharu
Nakayama
*a,
Yusuke
Fujii
a,
Kotaro
Fujimoto
a,
Makoto
Yoshimoto
a,
Aya
Kaide
b,
Takashi
Saeki
b and
Hironori
Asada
c
aMaterials Chemistry, Graduate School of Science and Engineering, Yamaguchi University, 2-16-1 Tokiwadai, Ube 755-8611, Japan. E-mail: nkymm@yamaguchi-u.ac.jp
bEnvironmental Science and Engineering, Graduate School of Science and Engineering, Yamaguchi University, 2-16-1 Tokiwadai, Ube 755-8611, Japan
cElectric Energy and Power Devices Engineering, Graduate School of Science and Engineering, Yamaguchi University, 2-16-1 Tokiwadai, Ube 755-8611, Japan
First published on 25th February 2016
Abstract
A new electrochemical process yielded a thin film consisting of stacked graphene (GR) sheets and manganese oxide. The process involved anodic deposition of manganese oxide nuclei on an electrode substrate and on the surface of GR sheets through electrolysis of aqueous Mn2+ ions in the presence of cationic GR colloids modified with poly(diallydimethylammonium) (PDDA+), followed by extraction of the PDDA. The catalytic activity of Mn oxide for the oxygen evolution reaction in an alkaline electrolyte was dramatically enhanced by hybridizing it with GR; an onset overpotential of 335 mV and a Tafel slope of 58 mV per decade were observed with excellent stability. These results can be attributed to a synergistic effect of the catalytic activity of nanosized Mn oxide deposits and the ability of GR to transfer electrons from the catalysts.
Introduction
Finding alternatives to fossil fuels is critical to maintaining energy resources and avoiding global climate change. Among the various energy storage options, hydrogen is regarded as the most promising energy carrier because it never discharges carbon dioxide as a combustion product. Since the energy from most renewable energy systems can be converted into electricity, electrochemical water splitting is an ideal process for producing hydrogen. However, the sluggish kinetics of the oxygen evolution reaction (OER, 2H2O → 4H+ + O2 + 4e−) that occurs at the counter electrode requires a considerable overpotential and thus limits the overall efficiency of water splitting. This has motivated the development of efficient catalysts for OER. Among these, ruthenium and iridium oxides are the best electrocatalysts in terms of activity and stability;1,2 however, their high cost and scarcity has hindered their use in large-scale applications.A highly efficient oxygen-evolving catalyst is involved in photosystem II (PS-II), which has a distorted chair structure composed of manganese and calcium (Mn4CaO5 cluster).3 Inspired by the high OER efficiency of this biological system, many research groups have focused on nanosized manganese oxides and derivatives thereof. Mn oxides are scalable catalysts because of their low cost, environmental friendliness, and natural abundance. However, their bulk structures, such as amorphous or β- and δ-MnO2, do not show high catalytic activity toward OER.4,5 This is because the poor electrical conductivity of MnO2 (10−5 to 10−6 S cm−1) can severely restrict the activity of catalytic systems involving electron transfer. The situation could be partly remedied by introducing a nanostructure, and a further improvement was achieved by using gold.5–9 These studies suggest that charge transfer from the transition metal oxides is critical to stabilize the active oxidation state of the catalysts.5 Thus, we would expect to obtain a similar effect with the use of graphene (GR), a two-dimensional sp2 nanocarbon with high surface area, electrical conductivity (107 to 108 S cm−1 for isolated single sheet),10 mechanical strength, and chemical stability. Surprisingly, however, only a few studies have been carried out on the OER properties of hybrid materials consisting of nanocarbons and transition metal oxides/hydroxides,11–14 despite their widespread use as electrode materials in supercapacitors. Tang et al. reported a small onset overpotential (330 mV) with the use of GR-supported ZnCo layered double hydroxide in 0.1 M KOH solution, where the catalyst was fabricated by coprecipitation of GR and ZnCO–LDH sheets and then mixed with Nafion to cast on the electrode.11 Phihusut et al. synthesized GR-supported CoC2O4 by mixing Co(NH3) and oxalic acid while the reduced GO was dispersed.12 Yuan et al. were the first to report GR-supported MnO2 nanowires as the OER catalyst; they grew these nanowires through the well-known redox reaction between MnO4− ions and GR.13 Electrodeposition is an ideal binder-free technique for fabricating transition metal oxides under ambient conditions and is particularly suited for the formation of uniform films even on substrates of complex shape. To the best of our knowledge, however, a one-step electrodeposition technique has never been attempted to construct hybrid materials of transition metal oxides and GR.
In 2004, we presented an electrochemical route to producing layered Mn oxides intercalated with alkaline metals and alkylammonium ions in thin film form, where aqueous Mn2+ ions were oxidized in the presence of the corresponding guest cations dissolved in water.15,16 In the present study, this strategy was extended to a colloidal species: cationic GR sheets. Specifically, Mn2+ ions were electrolyzed in a dispersion of cationic GR colloids, without any other cationic species, leading to restacking of the dispersed GR sheets. The process involved the anodic formation of negatively charged Mn oxide nuclei on the electrode substrate, which triggered the assembly of cationic GR colloids to the negatively charged oxide for charge compensation, followed by the deposition of more Mn oxide onto the deposited GR surface with excellent conductivity. The resulting thin film of GR-supported Mn oxide exhibited superior OER activity compared to pristine Mn oxide.
Experimental
Preparation of MnOx/GR film
Graphene oxide (GO) was prepared from commercial graphite powder (Z-5F, ITO GRAPHITE Co., Ltd.; BET surface area 13 m2 g−1) by a modified Hummers method.17 Cationic GR colloids were synthesized as described in the literature.18 In brief, an aqueous dispersion of GO (30 mL, 1.57 mg mL−1) was added to 100 mL of a solution of 0.5 wt% poly(diallydimethyl ammonium chloride) (PDDA+Cl−; Aldrich, Mw = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–500000) in water, and the dispersion was stirred overnight at room temperature. Then, 100 mg of NaBH4 was added, and the solution was stirred for 30 min, followed by refluxing for 24 h. The obtained GR colloids, whose surfaces were modified with PDDA+, were washed thoroughly in water and centrifuged repeatedly to completely remove any other cationic species. Zeta-potential measurements of the colloidal suspensions were carried out by the laser-Doppler method, using an ELSZ-2plus instrument (Otsuka Electronics) equipped with a 660 nm laser. The pH of the colloidal suspensions was adjusted by using a NaOH or HNO3 solution.
000–500000) in water, and the dispersion was stirred overnight at room temperature. Then, 100 mg of NaBH4 was added, and the solution was stirred for 30 min, followed by refluxing for 24 h. The obtained GR colloids, whose surfaces were modified with PDDA+, were washed thoroughly in water and centrifuged repeatedly to completely remove any other cationic species. Zeta-potential measurements of the colloidal suspensions were carried out by the laser-Doppler method, using an ELSZ-2plus instrument (Otsuka Electronics) equipped with a 660 nm laser. The pH of the colloidal suspensions was adjusted by using a NaOH or HNO3 solution.
All electrochemical experiments were conducted at room temperature using a Bio-logic SP-300 potentiostat/galvanostat. Electrolytes were prepared with doubly distilled water and deoxygenated by bubbling with purified nitrogen gas for at least 20 min prior to use. A fluorine-doped tin oxide (FTO)-coated glass slide (R = 10 Ω cm) with an active area of 1.8 × 1.0 cm2 was used as the working electrode on which the films were fabricated. Prior to electrodeposition, the electrode surface was ultrasonically cleaned in a mixed solution of ethanol and water and then rinsed thoroughly with water. Electrodeposition was made in a 50 mL aqueous dispersion containing PDDA+-modified GR colloids (1.48 × 10−2 mg mL−1) and 2 mM MnSO4·5H2O (99.9%, Wako Pure Chemicals). Electrolysis was conducted in the galvanostatic mode at an anodic current density of 0.06 mA cm−2 in a two-electrode system with a Pt mesh counter electrode, in order to avoid any contamination with cations from the reference electrode. The charge delivered during the electrolysis was 200 mC cm−2. The resulting film on the FTO electrode was rinsed thoroughly with water. As will be described later, the PDDA attached to the GR surface could be extracted by immersing the as-deposited film in an aqueous solution containing sodium polystyrene sulfonate (NaPSS; Aldrich, MW = 70000). For comparison, the same electrolysis run was performed in homogeneous solutions of 2 mM MnSO4 with 50 mM KCl and PDDACl, instead of cationic GR colloids, which yielded birnessite-type layered MnO2 intercalated with K+ and PDDA+ ions, respectively, as reported in our previous papers.15,16 These products will hereafter be denoted as K-Bir and PDDA-Bir, respectively.
Characterization
X-ray diffraction (XRD) patterns were recorded on a Rigaku Ultima IV diffractometer, using Cu Kα radiation (λ = 0.154051 nm). BET surface area was measured by nitrogen adsorption isotherm at 77 K using on a Gemini 2375 apparatus (Micromeritics). Field emission scanning electron microscopy (FE-SEM) data were obtained with a Hitachi S-4700Y SEM operating at 10 kV. Cross-sectional transmission electron microscopy (TEM) was conducted using a JEOL JEM-2010F microscope at an accelerating voltage of 200 kV. X-ray photoelectron spectra (XPS) were collected using a Thermo Scientific K-Alpha spectrometer with an AlKα (1486.6 eV) monochromatic source (1805 V, 3 mA). Wide- and narrow-range spectra were acquired with a pass energy of 50 eV and channel widths of 1.0 and 0.1 eV, respectively. The binding energy scale was calibrated with respect to the C 1s (284.8 eV) signal. Semiquantitative estimates of the relative atomic concentrations were obtained from the peak area ratios by considering the sensitivity factors provided by the instrument software.Electrical resistance (R in Ω) was measured for the electrodeposited films by the standard four-probe method with a HP34401A digital multimeter in the thickness direction of the specimen. In the measurement, Pt was sputtered onto the measuring point with a geometric area of 4 × 4 mm2 to make a good electrical contact. The resistance of the underlying FTO layer was as low as negligible. The resistivity (ρ in Ω cm) was calculated by the formula ρ = (S/t)R, where t is the film thickness and was measured to be ∼0.6 μm based on SEM measurements and S the Pt-sputtered area. Conductivity is the inverse of resistivity.
Electrochemical testing
Electrochemical testing was performed in a typical three-electrode cell with an electrolyte solution of 0.1 M KOH. A Pt mesh was used as the counter electrode, and a standard Ag/AgCl electrode (in saturated KCl) was used as the reference electrode. Linear sweep voltammetry (LSV) was performed at a scan rate of 1 mV s−1. Tafel plots were obtained from the rising part of the LSV curves. All potentials were calibrated to the reversible hydrogen electrode (RHE); i.e., E(RHE) = E(Ag/AgCl) + 0.059pH + 0.199. The onset potential for OER was estimated on the basis of the beginning of the linear portion of the Tafel plot. Chronoamperometry and chronopotentiometry were employed to further characterize the electrocatalysts in terms of turnover frequency (TOF) and stability, respectively. It is reported that the use of high anodic potential (>2.7 vs. RHE at pH 13) in the presence of O2 can cause corrosion of carbon electrode accompanied by CO2 evolution,19 which is not the case in our present study where the applied potential is far below 2.7 V.Results and discussion
The zeta-potential profile of the colloidal product obtained by reducing GO in the presence of PDDA is shown in Fig. 1a, along with that of GO for comparison. In the measured pH range, GO was charged negatively, owing to the ionization of functional groups.20,21 In contrast, the reduced product exhibited positive values at all pHs, confirming the cationic characteristics due to PDDA being anchored on the GR surface through π–π interactions.20,22 The cationic GR obtained in colloidal form will hereafter be denoted as PDDA-GR+. BET surface area of dried PDDA-GR+ powder was measured to be 7 m2 g−1 which was much smaller than that (436 m2 g−1) of the GR powder synthesized under similar conditions in the absence of PDDA+. This suggests a full coverage of the GR surface with PDDA, in good agreement with the positive zeta potential of PDDA-GR+ colloids. As shown in Fig. 1b, the film electrodeposited with PDDA-GR+ appeared more black, rather than brown like the MnO2 film prepared similarly with K+ (K-Bir) instead of PDDA-GR+, which was reflected in an increase in absorption over the whole wavelength region. Involvement of Mn in the film can be confirmed by the broad d–d* absorptions lying between 400 and 600 nm.23 It should be noted that the absorption curve remained unchanged after extracting PDDA+ by immersing the film in NaPSS solution (dashed line). Fig. 1c shows a cross-sectional TEM image of the film prepared with PDDA-GR+. A striped pattern with a periodicity of about 0.7 nm is seen, whose periodic structure evolves over a long distance. Close examination of the white stripes in the inset reveals another periodicity of about 0.2 nm, which is attributable to the lattice fringes corresponding to the 110 plane of GR.24 This strongly suggests that the cationic GR sheets were stacked and deposited onto the electrode substrate during the electrolysis of Mn2+. As will be discussed later, we assume that Mn oxide nuclei (or clusters) were deposited between the GR sheets. This was supported by energy-dispersive X-ray spectrometry (EDS) mapping analysis, which revealed that Mn, O, and C were present across the whole cross-section of the film (Fig. S1, ESI†).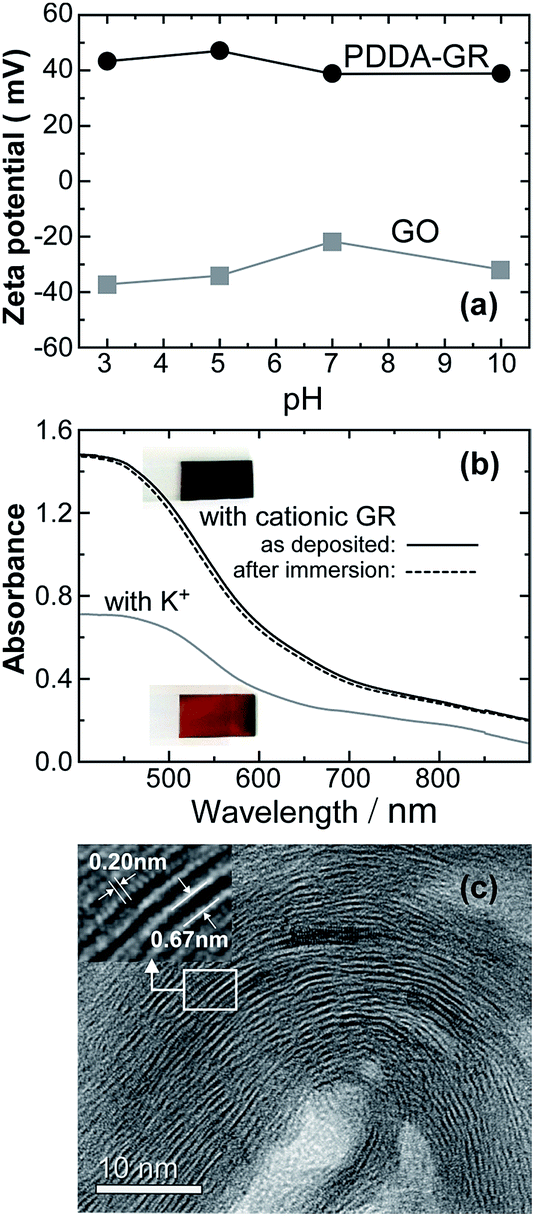 | ||
| Fig. 1 (a) Zeta potential profiles of GO and reduced GO in the presence of PDDA; (b) absorption spectra of the electrodeposited films with PDDA-GR+ and K+ and absorption spectra obtained after immersion of the former film in NaPSS solution; (c) cross-sectional TEM image of the electrodeposited film with PDDA-GR+. | ||
XRD patterns were recorded for the as-deposited film formed by the electrolysis of aqueous Mn2+ in the presence of PDDA-GR+ colloids, as well as for graphite, GO, and PDDA-GR+ samples in powder form (Fig. S2, ESI†). GO exhibited a sharp characteristic peak at 2θ = 9.46°, corresponding to a d-spacing of 0.93 nm.25 It shifted to 5.86° (a d-spacing of 1.51 nm) for PDDA-GR+, suggesting an expansion of the distance between the GR sheets, presumably due to the repulsion between the positive charges arising from the PDDA+ on the GR sheets.20 In the electrodeposited film, the lowest angle peak was observed at 11.6°, which is higher than that for PDDA-GR+, and no diffraction peaks due to PDDA-GR+ could be seen. This may indicate that the negatively charged Mn oxide brought the cationic GR sheets closer. A similar peak was reported by Zhang et al. for a hybrid material of MnO2 nanosheets and PDDA-GR+ sheets that was fabricated by the electrostatic coprecipitation method.20 The present electrodeposited film will hereafter be denoted as MnOx/PDDA-GR+.
XPS was used to characterize the MnOx/PDDA-GR+ film before and after immersion in NaCl/ethanol and aqueous NaPSS solutions (Fig. 2). The energy separation (ΔE) of the doublet peaks in the Mn 3s region is sensitive to the oxidation state of Mn in the oxide. The observed ΔE value (4.89) corresponds to an average oxidation state of 3.65 according to a linear relationship between the oxidation state of Mn and ΔE reported in the literature.26 The observed oxidation level (3.65) suggests that Mn in the oxide is in a mixed valence state between Mn4+ and Mn3+. Due to the involvement of Mn3+, the Mn oxide is negatively charged. Thus, it is reasonable to assume that PDDA-GR+ colloids accumulated on the negative charges resulting from the Mn3+ in the oxide. This is basically the same as our previous studies with soluble cations to form layered Mn oxides, where cations (C+) in solution segregate toward negative charges on Mn oxide grown anodically, as expressed by the following reaction:27
| Mn2+ + 2H2O + xC+ → CxMnx3+Mn1−x4+O2 + 4H+ + (2 − x)e− |
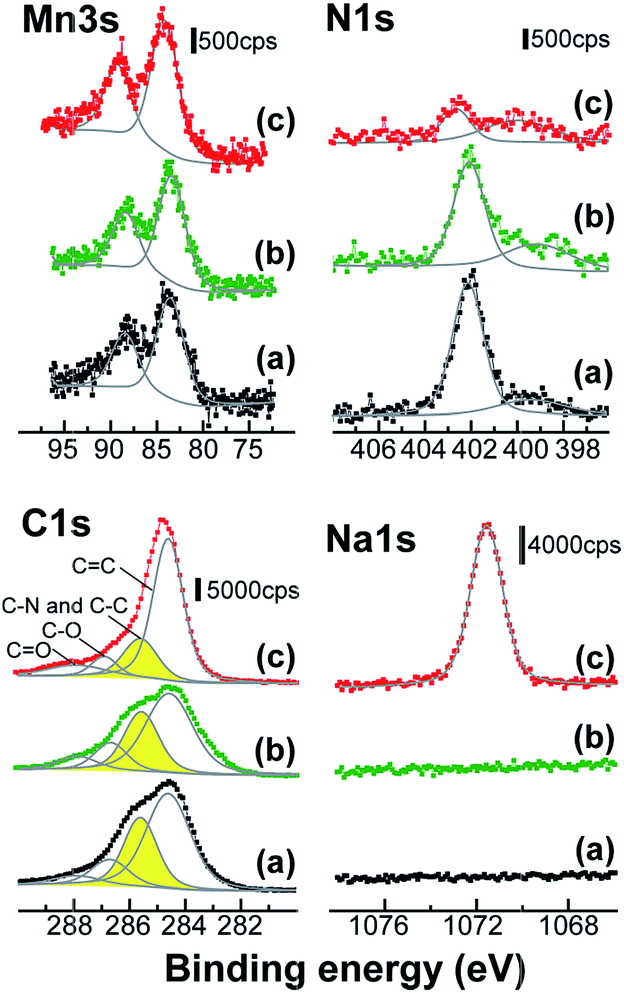 | ||
| Fig. 2 XPS spectra of the electrodeposited film with PDDA-GR+. The spectra were taken (a) before and after immersion in (b) NaCl/ethanol and (c) NaPSS solution. | ||
No significant change was observed in the oxidation level of Mn after immersion in a NaCl/ethanol or NaPSS solution. In the C 1s spectra, the peak at 285.6 eV was attributed to the carbon bound to the nitrogen belonging in PDDA.22 This peak was superimposed on the typical peaks arising from the four different types of carbon atoms in GR: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.6 eV), C–C (285.6 eV), C–O (286.7 eV), and CO (287.9 eV).28 The 285.6 eV peak due to PDDA decreased after immersion in NaPSS solution, while no significant change occurred with NaCl/ethanol. The disappearance of PDDA was confirmed by the dramatic decrease in intensity of the N 1s peaks, i.e., the principal peak at 402.1–402.6 due to N+ and a second component at 399.1–400.0 eV due to uncharged side species.22 Thus, the PDDA+ that had been attached to the GR surface was extracted into solution by NaPSS. On the basis of the areas of C 1s and Mn 2p1/2 peaks and their sensitivity factors, the atomic ratio of C/Mn in the film was calculated to be 10/1 in the as-deposited state, which decreased to 4.9/1 after immersion in NaPSS solution. Moreover, a Na 1s peak appeared after immersion in NaPSS solution. This demonstrates the incorporation of Na+ ions into the film from solution to maintain the charge neutrality after PDDA+ was expelled.
C (284.6 eV), C–C (285.6 eV), C–O (286.7 eV), and CO (287.9 eV).28 The 285.6 eV peak due to PDDA decreased after immersion in NaPSS solution, while no significant change occurred with NaCl/ethanol. The disappearance of PDDA was confirmed by the dramatic decrease in intensity of the N 1s peaks, i.e., the principal peak at 402.1–402.6 due to N+ and a second component at 399.1–400.0 eV due to uncharged side species.22 Thus, the PDDA+ that had been attached to the GR surface was extracted into solution by NaPSS. On the basis of the areas of C 1s and Mn 2p1/2 peaks and their sensitivity factors, the atomic ratio of C/Mn in the film was calculated to be 10/1 in the as-deposited state, which decreased to 4.9/1 after immersion in NaPSS solution. Moreover, a Na 1s peak appeared after immersion in NaPSS solution. This demonstrates the incorporation of Na+ ions into the film from solution to maintain the charge neutrality after PDDA+ was expelled.
The above-mentioned processes are represented in Scheme 1. In the first step, anodic oxidation of Mn2+ leads to nucleation of Mn oxide with a mixed oxidation state of Mn4+/Mn3+. The surface of this Mn oxide is charged negatively owing to Mn3+. The negative charges on the Mn deposits are neutralized by the accumulation of cationic PDDA-GR+ colloids because no cationic species exist in the bath except for Mn2+. The deposited PDDA-GR+ sheets can then act as a substrate for subsequent electrodeposition of Mn oxide because GR is an excellent conductor. This cycle is repeated during electrolysis, producing a multilayered nanohybrid film of MnOx/PDDA-GR+. The PDDA+ anchored on the surface of the GR colloids can be extracted into solution by PSS−. At the same time, the Na+ ions in solution are incorporated into the film or onto the GR surface for charge compensation. The resulting end product will hereafter be denoted as MnOx/GR.
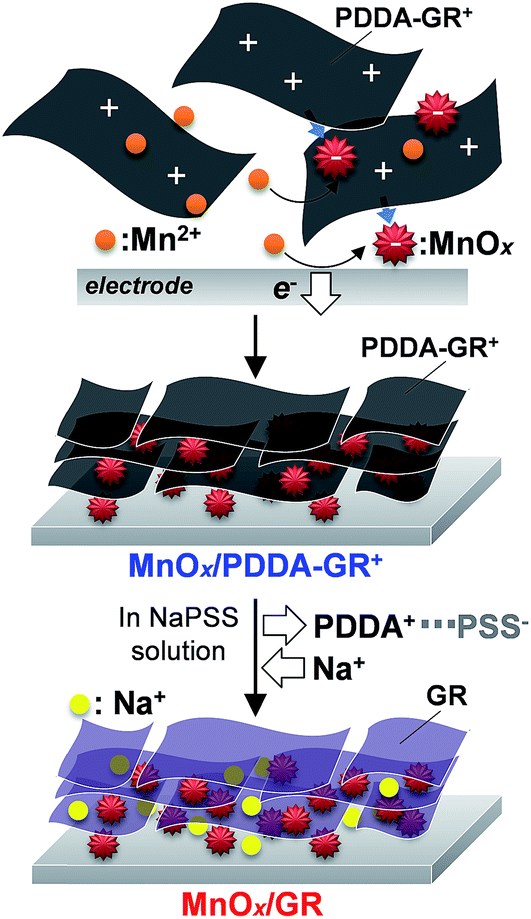 | ||
| Scheme 1 Schematic of the synthesis of MnOx/GR film. | ||
The film growth accompanying the incorporation of the highly conducting GR was also indicated by a decreasing voltage profile during electrodeposition, not observed in the presence of other cationic species (Fig. S3, ESI†). Indeed, electrical conductivity in the thickness direction was examined for the electrodeposited films on FTO by using the standard four-probe resistance measurement. The obtained results are summarized in Table 1. The films with GR were higher in electrical conductivity than pure birnessite films. A further increase was achieved by the extraction of insulating PDDA.
| Sample | MnOx/GR | MnOx/PDDA-GR+ | PDDA-Bir | K-Bir |
| Conductivity (S cm−1) | 9.3 × 10−4 | 1.0 × 10−5 | 7.3 × 10−6 | 8.3 × 10−6 |
Fig. 3a presents linear sweep voltammograms (LSV) of GR-supported Mn oxide electrodes in 0.1 M KOH solution, obtained at a scan rate of 1 mV s−1, along with those of K-Bir and PDDA-Bir films. Here, the charge delivered during film deposition was fixed at 200 mC cm−2. The corresponding Tafel plots are shown in Fig. 3b. The onset potential was determined on the basis of the beginning of the linear portion in the plots. MnOx/GR and MnOx/PDDA-GR+ electrodes exhibited onset potentials of 1.56 and 1.58 V vs. RHE, respectively, which was about 200 mV less positive than those of the electrodes without GR: 1.76 V for both K-Bir and PDDA-Bir. Since GR has no catalytic activity for OER,11,12 clearly, the incorporated GR effectively enhanced the OER ability of Mn oxide. MnOx/GR showed a significantly smaller Tafel slope (58 mV dec−1) than MnOx/PDDA-GR+ (89.0 mV dec−1), K-Bir (112 mV dec−1), and PDDA-Bir (100 mV dec−1). Furthermore, 58 mV dec−1 is better than most values reported for pure Mn oxides4,29,30 and comparable to the values reported for Au-anchored MnO2 nanowires (51–68 mV dec−1).9
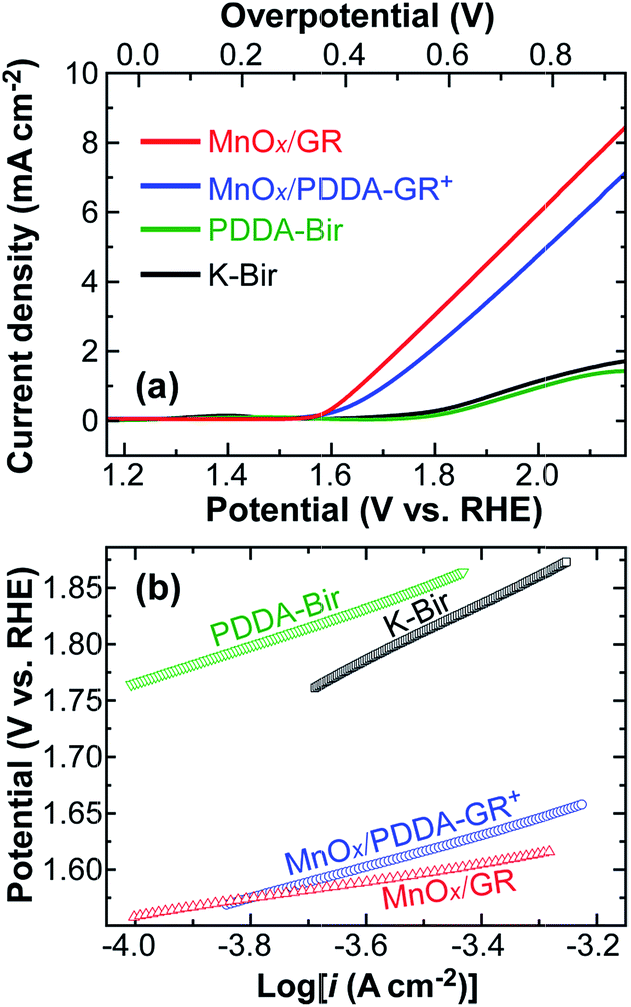 | ||
| Fig. 3 (a) LSVs for the indicated films in 0.1 M KOH solution, obtained at a scan rate of 1 mV s−1; (b) the corresponding Tafel plots. | ||
Chronoamperometry was applied to further examine the catalytic ability of GR-supported and unsupported Mn oxides. The results are shown in Fig. 4a. At all potential steps, the GR-supported Mn oxides exhibited much larger current responses than K-Bir. Note that the difference between MnOx/GR and MnOx/PDDA-GR+ diminished with increasing applied potential. This behavior can be ascribed to the detachment of PDDA cations by anodic polarization. A detailed investigation is now underway. The significant current decrease observed for K-Bir during each step is presumably due to the instability of the catalyst. The turnover frequency (TOF) per Mn atom was taken at 1.68 V (η = 0.45 V) for comparison, because several research groups have employed this condition for obtaining TOF.4,29,30 We calculated TOF via the equation13,31
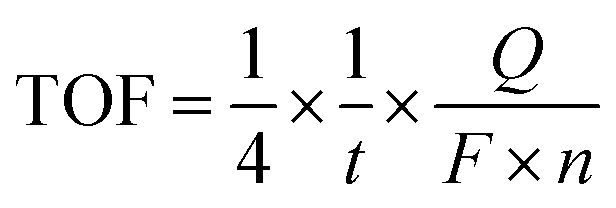
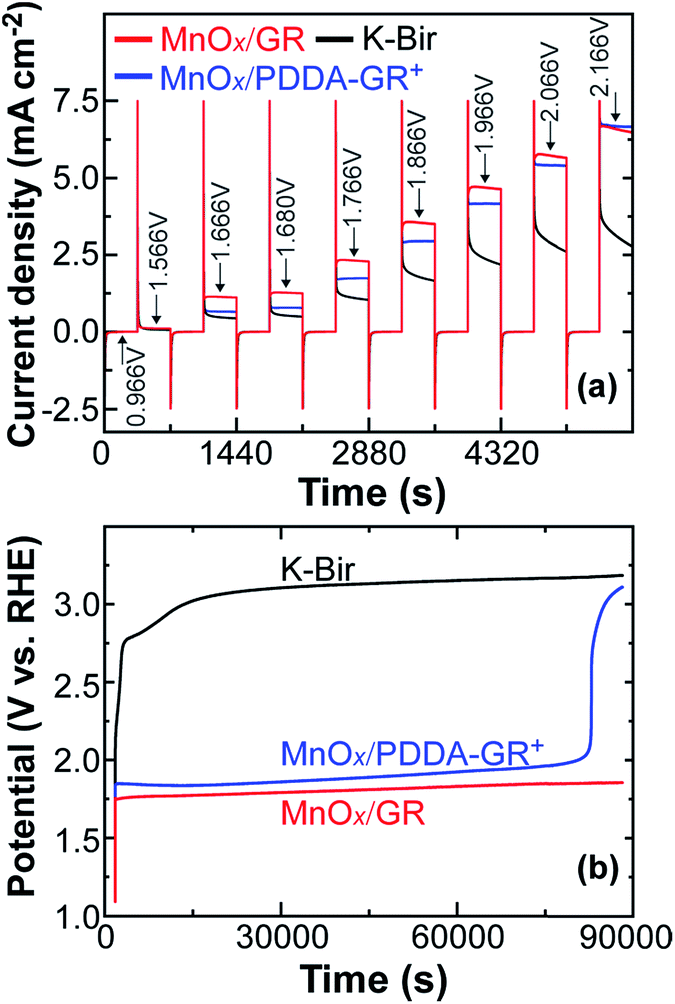 | ||
| Fig. 4 (a) Chronoamperometric curves for the indicated films under a series of applied potentials in 0.1 M KOH solution; (b) chronopotentiometric curves of the indicated films under a constant anodic current density of 2.5 mA cm−2. | ||
The stability of catalysts was evaluated by chronopotentiometry with an applied anodic current density of 2.5 mA cm−2 (Fig. 4b). The potential of the K-Bir electrode rapidly reached the potential region where OER can take place even at the underlying FTO surface, the accompanying formation of O2 bubbles causing the film to detach. As expected from Fig. 4a, the observed potential for MnOx/PDDA-GR+ was somewhat positive and increased gradually up to the abrupt rise at 82900 s. The best stability was observed for MnOx/GR, where the increase in potential remained within 0.11 V (1.748–1.856 V). Another stability test was carried out in CV mode using a grassy carbon rotating disk electrode. A stable response was observed for MnOx/GR until 1000 cycles (Fig. S4, ESI†).
Conclusions
We developed a new electrochemical technique for fabricating nanohybrid films composed of stacked graphene sheets and incorporated Mn oxide deposits. The catalytic activity of Mn oxide for OER was dramatically enhanced through hybridization with GR, which also resulted in excellent stability. The observed Tafel slope (58 mV dec−1) was better than most values reported to date for Mn oxide-based materials and comparable to those of Au-anchored MnO2 nanowires. This can be attributed to a synergistic effect between the catalytic activity of nanosized Mn oxide and the electron transferring ability of GR. In the future study, the catalyst film will be deposited on a 3D porous substrate with high surface area to enhance the catalytic efficiency.Acknowledgements
We gratefully acknowledge the financial support of the Yamaguchi Industrial Promotion Foundation and Yamaguchi University Opt-Energy Research Center.Notes and references
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- Y. Zhang, C. Wang, N. Wan and Z. Mao, Int. J. Hydrogen Energy, 2007, 32, 400–404 CrossRef CAS.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- L. C. Seitz, T. J. P. Hersbach, D. Nordlund and T. F. Jaramillo, J. Phys. Chem. Lett., 2015, 6, 4178–4183 CrossRef CAS PubMed.
- M. S. El-Deab, M. I. Awad, A. M. Mohammad and T. Ohsaka, Electrochem. Commun., 2007, 9, 2082–2087 CrossRef CAS.
- A. M. Mohammad, M. I. Awad, M. S. El-Deab, T. Okajima and T. Ohsaka, Electrochim. Acta, 2008, 53, 4351–4358 CrossRef CAS.
- Y. Gorlin, C.-J. Chung, J. D. Benck, D. Nordlund, L. Seitz, T.-C. Weng, D. Sokaras, B. M. Clemens and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 4920–4926 CrossRef CAS PubMed.
- H. Zhang, C. Lin, F. Du, Y. Zhao, P. Gao, H. Chen, Z. Jiao, X. Li, T. Zhao and Y. Sun, ACS Sustainable Chem. Eng., 2015, 3, 2049–2057 CrossRef CAS.
- B. Marinho, M. Ghislandi, E. Tkalya, C. E. Koning and G. With, Powder Technol., 2012, 221, 351–358 CrossRef CAS.
- D. Tang, Y. Han, W. Ji, S. Qiao, X. Zhou, R. Liu, X. Han, H. Huang, Y. Liu and Z. Kang, Dalton Trans., 2014, 43, 15119–15125 RSC.
- D. Phihusut, J. D. Ocon, B. Jeong, J. W. Kim, J. K. Lee and J. Lee, Electrochim. Acta, 2014, 140, 404–411 CrossRef CAS.
- W. Yuan, P. K. Shen and S. P. Jiang, J. Mater. Chem. A, 2014, 2, 123–129 CAS.
- N. I. Andersen, A. Serov and P. Atanassov, Appl. Catal., B, 2015, 163, 623–627 CrossRef CAS.
- M. Nakayama, S. Konishi, A. Tanaka and K. Ogura, Chem. Lett., 2004, 33, 670–671 CrossRef CAS.
- M. Nakayama, H. Tagashira, S. Konishi and K. Ogura, Inorg. Chem., 2004, 43, 8215–8217 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Wang, D. Yu, L. Dai, D.-W. Chang and J.-B. Beak, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- H. Liu, Q. Xu, C. Yan and Y. Qiao, Electrochim. Acta, 2011, 56, 8783–8790 CrossRef CAS.
- J. Zhang, J. Jiang and X. S. Zhao, J. Phys. Chem. C, 2011, 115, 6448–6454 CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- D.-Q. Yang, J.-F. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481–4484 CrossRef CAS PubMed.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Electrochem. Soc., 2005, 152, E384–E389 CrossRef CAS.
- X. Jin, K. Adpakpang, I. Y. Kim, S. M. Oh, N.-S. Lee and S.-J. Hwang, Sci. Rep., 2015, 5, 11057 CrossRef PubMed.
- C. Hontoria-Lucas, A. J. López-Peinado, J. d. D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef CAS.
- M. Nakayama, S. Konishi, H. Tagashira and K. Ogura, Langmuir, 2005, 21, 354–359 CrossRef CAS PubMed.
- S. Stankovich, D. Dikin, R. D. Piner, K. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- M. Fekete, R. K. Hocking, S. L. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 CAS.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 CAS.
- L. Tong, M. Göthelid and L. Sun, Chem. Commun., 2012, 48, 10025–10027 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: EDS elemental mapping and XRD pattern of MnOx/PDDA-GR+ film, potential–time curves for the electrolysis, and stability test with RDE. See DOI: 10.1039/c6ra00255b |
| This journal is © The Royal Society of Chemistry 2016 |