
Synthesis of 1,3,4-oxadiazoles as promising anticoagulant agents
Abstract
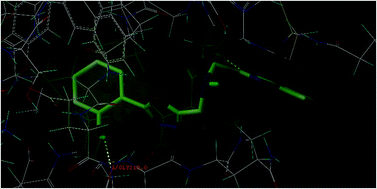
In the present study, a series of 2,5-disubstituted-1,3,4-oxadiazole derivatives (4a–4k) were designed and subjected to molecular docking simulation studies onto the enzymes vitamin K epoxide reductase (PDB: 3KP9) and factor Xa (PDB: 1NFY) to visualize their binding affinity towards the said target proteins. In silico ADME studies highlighted that the designed compounds are safe enough and have the potential to be considered as drug like molecules, indicating the appreciable ADME property & probable toxicity of the designed compounds. The title compounds were synthesized from (benzo[d]oxazol-2-yl)methanamine and evaluated for in vitro radical scavenging properties and ex vivo anticoagulant activity. The results of the ex vivo anticoagulant evaluation highlighted that the compounds exhibited a significant increase in prothrombin time and clotting time, and a minimal increase in the activated partial thromboplastin time, indicating that the compounds can be considered for promising anticoagulant therapy. The results of the radical scavenging experiments indicated that the compounds have substantial antioxidant activity.
 Please wait while we load your content...
Please wait while we load your content...

