
Formation of calcium phosphate nanostructures under the influence of self-assembling hybrid elastin-like-statherin recombinamers†

Abstract
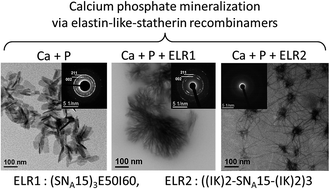
The self-assembling properties of thermally-sensitive amphiphilic elastin-like multiblock recombinamers have been combined with the capacities of calcium phosphate binding of the SNA15 epitope inspired by the salivary protein statherin. In this regard, the interaction between calcium and phosphate ions was examined in the presence of two hybrid recombinamers. The first recombinamer comprised a simple amphiphilic diblock in which the SNA15 epitopes were combined, at the gene level, to the hydrophilic end. This recombinamer can self-assemble into nanoparticles that can control the transformation of amorphous calcium phosphate (ACP) into a fibre-like hydroxyapatite structure. In the other recombinamer, the SNA15 domains are distributed along the monomer chain, with the hydrophilic blocks being distributed amongst the hydrophobic ones. In this case, the resulting nanohybrid ACP/recombinamer organises into neuron-like structures. Thus, combining the amphiphilic elastin-like recombinamers to the SNA15 functionality is a powerful mean to tune the formation of different complex calcium phosphate nanostructures.
 Please wait while we load your content...
Please wait while we load your content...

