
A solution to achieve good reusability of MNPs Fe3O4-supported (S)-diphenylprolinoltrimethylsilyl ether catalysts in asymmetric Michael reactions†
Tao Wua,
Dandan Fenga,
Bing Xie*b and
Xuebing Ma*a
aKey Laboratory of Applied Chemistry of Chongqing Municipality, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, P. R. China. E-mail: zcj123@swu.edu.cn; Fax: +86 23 68253237; Tel: +86 23 68253237
bSchool of Chemistry and Environmental Science, Guizhou Minzhu University, Guiyang, 550025, P. R. China. E-mail: bing_xie1963@hotmail.com; Fax: +86 851 3610278; Tel: +86 851 3610278
First published on 26th February 2016
Abstract
A new supported (S)-diphenylprolinol trimethylsilyl ether (Fe3O4/PVP@SiO2/ProTMS) using polyvinylpyrrolidone (PVP)-modified MNPs Fe3O4 as a support was prepared via one-pot surface-modification, and exhibited high yield (93–99%), excellent diastereoselectivities (syn/anti = 81–96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19–4) and enantioselectivities (95–98% ee) in the asymmetric Michael addition of propanal to various nitroalkenes. TGA, XRD, IR, SEM, TEM, elemental analysis and N2 adsorption–desorption isotherm demonstrated that the adsorption of PVP onto MNPs Fe3O4 resulted in the spectacular change in the chemical composition, surface morphology and pore structure of Fe3O4/PVP@SiO2/ProTMS. The catalyst could be easily separated from the reaction by an external magnet and reused for ten times with high yields (77–99%) and unchanged excellent stereoselectivities (97–98% ee and syn/anti = 96/4) in the Michael addition for the first time.
:19–4) and enantioselectivities (95–98% ee) in the asymmetric Michael addition of propanal to various nitroalkenes. TGA, XRD, IR, SEM, TEM, elemental analysis and N2 adsorption–desorption isotherm demonstrated that the adsorption of PVP onto MNPs Fe3O4 resulted in the spectacular change in the chemical composition, surface morphology and pore structure of Fe3O4/PVP@SiO2/ProTMS. The catalyst could be easily separated from the reaction by an external magnet and reused for ten times with high yields (77–99%) and unchanged excellent stereoselectivities (97–98% ee and syn/anti = 96/4) in the Michael addition for the first time.
Introduction
The Jørgensen–Hayashi organocatalysts, i.e., α,α-diarylprolinoltrimethylsilyl ethers,1 which activate aldehydes via an enamine2 and α,β-unsaturated carbonyl compounds via an iminium ion,3 can be considered as one of the most famous leading actors among the versatile organocatalysts in the asymmetric reactions such as Aldol, Michael, cycloaddition, epoxidation and desymmetrization.4 Moreover, Jørgensen–Hayashi catalysts have been utilized in asymmetric tandem and multi-component reactions, through which large quantities of enantiopure compounds with multiple stereogenic carbon centers can be conveniently constructed.5 However, the usage of organocatalyst, typically 10–30 mol%, was needed to achieve desirable catalytic performance in most cases of organocatalyzed reactions. From the viewpoint of green chemistry, several efficient and economical protocols using polymer,6 inorganic material,7 ionic liquid8 and graphene oxide9 as catalyst supports were developed to achieve the reuse of expensive Jørgensen–Hayashi catalysts.On the other hand, due to the potential advantages of inexpensive, non-toxic, chemically stable properties and re-collection by simple magnetic decantation without filtration or centrifugation, the magnetic nanoparticles (MNPs) of Fe3O4 had emerged recently as a new type of catalyst support in heterogeneous asymmetric organocatalysis with good catalytic performances, especially in Aldol addition.10 However, in the asymmetric Michael addition of aldehyde to nitroalkene, all the reported MNPs Fe3O4-supported Jørgensen–Hayashi catalysts gave the unsatisfactory reusability. In 2010, W. Wang et al. developed Fe3O4@SiO2-supported Jørgensen–Hayashi catalyst, whose yield and stereoselectivity significantly decreased to 54%, syn/anti = 83:17 and 85% ee in the third run.11 One year later, A. Pericás et al. immobilized Jørgensen–Hayashi catalyst onto azide functionalized MNPs Fe3O4@SiO2. Unfortunately, the similar sharp drop in catalytic performance was found in the fourth run (57%, syn/anti = 72:28 and 92% ee).12 Recently, A. Ouali et al. also reported on a poly(benzylazide)styrene-functionalized magnetic Co/C-supported (S)-α,α-diphenylprolinoltrimethylsilyl ether with a sharp drop in the activity in the 3rd run.13 Untill now, the good reusability of MNPs-supported Jørgensen–Hayashi catalyst could not be achieved in the asymmetric Michael addition of aldehyde to nitroalkene.
In our attempts to solve the reusability of MNPs-supported Jørgensen–Hayashi catalyst, we report the supporting of (S)-α,α-diphenylprolinoltrimethylsilyl ether onto polyvinylpyrrolidone (PVP)-modified MNPs of Fe3O4 through the hydrolysis of –Si(OCH3)3 by the facile one-pot surface-modification (Scheme 1). Due to the vital function of PVP, the as-synthesized Fe3O4/PVP@SiO2/ProTMS possessed the excellent catalytic performances (93–99% yield, syn/anti = 81–96:19–4, 95–98% ee) and good reusability with the unchanged excellent stereoselectivities (syn/anti = 96:4, 98% ee) and moderate yield (82%), even in the 10th run.
 | ||
| Scheme 1 The one-pot preparation of PVP-modified MNPs Fe3O4-supported Jørgensen–Hayashi organocatalysts. | ||
Experimental
Characterization
1H and 13C NMR spectra were conducted using a Bruker av-600 NMR instruments, in which all chemical shifts were reported down-field in ppm relative to the hydrogen and carbon resonances of TMS. FT-IR spectroscopy was performed on a Perkin-Elmer model GX spectrometer using the KBr pellet. Thermogravimetry-differential thermal analysis was carried out on a SBTQ600 thermal analyzer at a heating rate of 10 °C min−1 from 40 to 800 °C using N2 as protective gas (100 mL min−1). Elemental analysis was performed using a vario Micro cube elemental analyzer instrument. The surface morphologies of the samples were observed by JSM-6510LV scanning electron microscopy and Tecnai G2 F20 transmission electron microscope, operated at 20 kV/15 mA and 200 kV respectively. X-ray powder diffraction patterns were detected on an XRD-7000 S/L instrument: Cu-Kα radiation, X-ray tube settings of 40 kV/30 mA and a step size of 2° min−1 in the 10–100° (2θ) range. N2 adsorption–desorption isotherm was carried out at 77.4 K using an Autosorb-1 apparatus (Quantachrome), in which the sample was degassed at 120 °C for 12 h before measurement, and the specific surface area and the pore diameter were calculated by the BET method and BJH model, respectively. Magnetization curve was determined by HH-15 vibrating sample magnetometer. The syn/anti ratios and % ee values of products were determined by 1H NMR and Agilent LC-1200 HPLC using Daicel Chiralpak OD-H 4.6 mm × 25 cm column under 20 °C, 220 nm and 1.0 mL min−1 conditions, respectively.Synthesis of Jørgensen–Hayashi catalyst 5
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.74 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.26–7.39 (m, 4H, Ar-H), 8.90 (s, 1H, COOH) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.7 (NCHCH2), 52.0 (NCH2), 57.9 (NCH), 71.0, 75.9, 80.7 (OCH2, OCH and OC(CH3)3), 114.0 (CH2), 126.3, 127.7, 127.8, 136.4, 137.2 (Ph, CH), 153.9 (NCO), 178.2 (CO) ppm.CH2), 5.74 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.26–7.39 (m, 4H, Ar-H), 8.90 (s, 1H, COOH) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.7 (NCHCH2), 51.3 (NCH2), 57.9 (NCH), 71.0, 75.9, 80.7 (OCH2, OCH and OC(CH3)3), 114.0 (CH2), 126.3, 127.7, 127.8, 136.4, 137.2 (Ph, CH), 153.9 (NCO), 178.2 (CO) ppm.CH2), 5.72 (d, J = 17.6 Hz, 1H, CH2), 6.69 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.16–7.49 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.5 (NCHCH2), 52.9 (NCH2), 65.1 (NCH), 70.6, 76.4, 80.7 (OCH2, OCH, OC(CH3)3), 81.6 (COH), 113.8 (CH2), 126.2, 127.1, 127.2, 127.5, 127.8, 127.9, 136.6, 137.1, 137.5 (Ph, CH), 145.7 (NCO) ppm.CH2), 5.73 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.13–7.57 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 33.0 (NCHCH2), 52.4 (NCH2), 63.4 (NCH), 70.5, 76.9, 79.5 (OCH2, OCH and OC(CH3)3), 113.8 (CH2), 125.4, 126.0, 126.3, 126.4, 126.6, 127.8, 128.0, 128.3, 136.5, 137.1, 137.9, 144.9, 147.3 (Ph, CH) ppm.CH2), 5.64 (d, J = 17.6 Hz, 1H, CH2), 6.61 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.10–7.39 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 2.1 (CH3), 34.3 (NCHCH2), 52.9 (NCH2), 63.7 (NCH), 70.5, 79.2, 82.9 (OCH2, OCH, OC(CH3)3), 113.7 (CH2), 126.2, 126.8, 126.9, 127.4, 127.5, 127.6, 127.8, 128.4, 136.5, 136.9, 138.1, 145.3, 146.6 (Ph, CH) ppm.
CH2), 5.74 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.26–7.39 (m, 4H, Ar-H), 8.90 (s, 1H, COOH) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.7 (NCHCH2), 52.0 (NCH2), 57.9 (NCH), 71.0, 75.9, 80.7 (OCH2, OCH and OC(CH3)3), 114.0 (CH2), 126.3, 127.7, 127.8, 136.4, 137.2 (Ph, CH), 153.9 (NCO), 178.2 (CO) ppm.CH2), 5.74 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.26–7.39 (m, 4H, Ar-H), 8.90 (s, 1H, COOH) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.7 (NCHCH2), 51.3 (NCH2), 57.9 (NCH), 71.0, 75.9, 80.7 (OCH2, OCH and OC(CH3)3), 114.0 (CH2), 126.3, 127.7, 127.8, 136.4, 137.2 (Ph, CH), 153.9 (NCO), 178.2 (CO) ppm.CH2), 5.72 (d, J = 17.6 Hz, 1H, CH2), 6.69 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.16–7.49 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 28.2 (CH3), 36.5 (NCHCH2), 52.9 (NCH2), 65.1 (NCH), 70.6, 76.4, 80.7 (OCH2, OCH, OC(CH3)3), 81.6 (COH), 113.8 (CH2), 126.2, 127.1, 127.2, 127.5, 127.8, 127.9, 136.6, 137.1, 137.5 (Ph, CH), 145.7 (NCO) ppm.CH2), 5.73 (d, J = 17.6 Hz, 1H, CH2), 6.70 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.13–7.57 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 33.0 (NCHCH2), 52.4 (NCH2), 63.4 (NCH), 70.5, 76.9, 79.5 (OCH2, OCH and OC(CH3)3), 113.8 (CH2), 125.4, 126.0, 126.3, 126.4, 126.6, 127.8, 128.0, 128.3, 136.5, 137.1, 137.9, 144.9, 147.3 (Ph, CH) ppm.CH2), 5.64 (d, J = 17.6 Hz, 1H, CH2), 6.61 (dd, J = 10.9, 17.6 Hz, 1H, CH), 7.10–7.39 (m, 14H, Ar-H) ppm; 13C NMR (151 MHz, CDCl3): δ 2.1 (CH3), 34.3 (NCHCH2), 52.9 (NCH2), 63.7 (NCH), 70.5, 79.2, 82.9 (OCH2, OCH, OC(CH3)3), 113.7 (CH2), 126.2, 126.8, 126.9, 127.4, 127.5, 127.6, 127.8, 128.4, 136.5, 136.9, 138.1, 145.3, 146.6 (Ph, CH) ppm.Preparation of MNPs Fe3O4 and Fe3O4/PVP
FeCl3·6H2O (11.0 g, 40.7 mmol) and FeCl2·4H2O (4.0 g, 20.4 mmol) in water (50 mL) were mixed well at room temperature under argon. The reaction mixture was heated to 85 °C and then added aqueous ammonia (23%) dropwise to a pH of 8–9. After being aged at 85 °C for 4 h, the MNPs Fe3O4 were separated by magnetic decantation and washed with water to pH = 7. Subsequently, MNPs Fe3O4 (4.0 g) were well-dispersed in water (50 mL) and added aqueous polyvinylpyrrolidone (PVP) solution (10.0 mL, 25 g L−1). After being stirred at room temperature for 12 h, the reaction mixture was added acetone (250 mL) and stirred for 10 min. The MNPs Fe3O4/PVP were obtained by magnetic decantation, washed with ethanol (50 mL × 2) and dried under vacuum at 55 °C for 24 h. Anal. calcd for Fe3O4/PVP found: C, 0.01; H, 0.23; N, 0.05.Preparation of supported Jørgensen–Hayashi catalysts Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS
A mixture of 3-mercaptotrimethoxysilane (MPTMS, 0.25 mL, 1.14 mmol), AIBN (41.0 mg, 0.25 mmol) and 5 (0.23 g, 0.5 mmol) in 15 mL of CHCl3 was stirred at 80 °C for 12 h under a N2 atmosphere and the solvent was removed under reduced pressure. The residues containing MPTMS and ProTMS were added slowly to ethanol solution containing Fe3O4 or Fe3O4/PVP (25 mL, 125.0 mg) pretreated with aqueous ammonia (0.75 mL, 23%) and tetraethyl orthosilicate (0.13 mL, TEOS) for 2 h. After the resulting mixture was stirred at room temperature for 24 h, the pale yellow MNPs-supported Jørgensen–Hayashi Fe3O4@SiO2/ProTMS (0.46 g) and Fe3O4/PVP@SiO2/ProTMS (0.57 g) catalysts were separated magnetically, washed with CHCl3 (25 mL × 3), ethanol (25 mL × 3) and dried under vacuum for 4 h. Anal. calcd for Fe3O4@SiO2/ProTMS found: C, 31.51; H, 4.10; N, 1.02; S, 6.15 and Fe3O4/PVP@SiO2/ProTMS found: C, 31.56; H, 3.22; N, 0.90; S, 5.42.General procedure for the Michael reaction
A reaction mixture of (E)-nitrostyrene (40.0 mg, 0.27 mmol), Fe3O4/PVP@SiO2/ProTMS (30.0 mg, 7 mol%) and CHCl3 (2.0 mL) was stirred at 0 °C for 10 min, added propanal (94.0 mg, 1.62 mmol) by syringe and monitored by TLC until completion. The catalyst was separated using an external magnetic field, washed with CHCl3 (2.0 mL × 3) and reused directly for the next catalytic cycle. The combined organic layers were concentrated and purified by silica gel column chromatography using petroleum ether/ethyl acetate (v/v = 5/1) as eluents to afford the Michael adduct. The diastereoselectivities (syn/anti) and enantioselectivities (% ee) of products were determined by 1H NMR and HPLC on a chiral stationary phase (Daicel Chiralpak OD-H, AD-H, AS-H or OJ-H, 4.6 mm × 25 cm column, see ESI†), respectively.Results and discussion
Preparation of MNPs-supported organocatalyst
As described in Scheme 2, Jørgensen–Hayashi catalyst 5 could be synthesized using Boc-L-hydroxyproline as starting material with the moderate to good yields in each step (77–95%).14 The TEM image showed that the spherical MNPs of Fe3O4 with the 20–30 nm diameters could be generated at a pH of 8–9 by simple co-precipitation of FeCl3 and FeCl2 with molar ratio of 2 in the presence of aqueous ammonia (23%). After being modified for 12 h at room temperature in aqueous PVP solution, TEM image revealed that MNPs Fe3O4/PVP had no significant change in the surface morphology and particle diameter. According to the content of nitrogen (0.05%) detected by elemental analysis, the loading of PVP onto the backbone of Fe3O4 was calculated to be very low (about 0.4%). | ||
| Scheme 2 The synthetic route to the Jørgensen–Hayashi organocatalyst 5. | ||
As described in Scheme 1, the Jørgensen–Hayashi catalyst 5 attached to ProTMS could be immobilized by one-pot to prepare the supported organocatalysts Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS through the radical addition of sulfhydryl (–SH) to CC double bond in 5 and subsequent surface-modification taking advantage of the hydrolysis of Si(OCH3)3 on the surface of MNPs Fe3O4 and Fe3O4/PVP. Based to the nitrogen contents by elemental analysis (1.02% and 0.90%), the loading capacities of Jørgensen–Hayashi catalyst 5 in Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS were calculated to be 0.73 mmol g−1 and 0.64 mmol g−1, respectively. Moreover, the loadings of MPTMS onto Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS were also determined by elemental analysis of sulphur (6.15% and 5.42%) to be 1.92 mmol g−1 and 1.69 mmol g−1. Owing to the partial radical addition of sulfhydryl (–SH) to CC double bond in 5, the free MPTMS were found to be 1.19 mmol g−1 and 1.05 mmol g−1. Based on the loaded MPTMS and ProTMS, it was found that Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS possessed the same molar ratio (1.6/1) of free MPTMS to ProTMS. Furthermore, the superparamagnetic behavior of Fe3O4@SiO2/ProTMS (21.5 emu g−1) and Fe3O4/PVP@SiO2/ProTMS (10.4 emu g−1) at 298 K was evidenced by the zero coercivity and resonance of each magnetization loop in the magnetization curves (Fig. 1).
 | ||
| Fig. 1 Magnetization curves for Fe3O4/PVP (a), Fe3O4@SiO2/ProTMS (b), 10th-recycled Fe3O4/PVP@SiO2/ProTMS (c) and Fe3O4/PVP@SiO2/ProTMS (d). | ||
The role of polyvinylpyrrolidone (PVP)
Poly(vinylpyrrolidone) (PVP, K29–32), amphiphilic and nonionic polymer, was widely used in science and technology, and could be adsorbed onto a broad range of different materials as stabilizing agent.15 Herein, we endeavoured to elucidate where PVP was adsorbed onto the backbone of MNPs Fe3O4 and what changes in chemical and physical properties were resulted from PVP.The covalent attachment of Jørgensen–Hayashi catalyst 5 to the backbone of Fe3O4 MNPs is clearly corroborated by FT-IR spectroscopy (Fig. 2), which confirmed by (1) the asymmetrical, symmetrical stretching vibration and flexural vibration of Si–O–Si at 1067, 807 and 459 cm−1,16 (2) the stretching vibrations of C–S and Si–C bonds at 1249 and 702 cm−1,17 (3) the various C–H stretching vibrations and characteristic phenyl ring at 3056, 3025, 2930 cm−1 and in the 1632–1448 cm−1 range, respectively. After being modified by PVP (0.4%), there was no significant difference in IR spectra between Fe3O4/PVP and bare Fe3O4. Furthermore, all Fe3O4/PVP@SiO2/ProTMS and Fe3O4@SiO2/ProTMS had a strong stretching vibration of C–O bond at 1134 cm−1, which implied that a large amount of –SiOCH3 moieties existed on the surface of Fe3O4/PVP@SiO2/ProTMS and Fe3O4@SiO2/ProTMS.
 | ||
| Fig. 2 IR spectra of Fe3O4 MNPs (a), Fe3O4/PVP MNPs (b), Jørgensen–Hayashi catalyst 5 (c), Fe3O4/PVP@SiO2/ProTMS (d) and Fe3O4@SiO2/ProTMS (e). | ||
The effect of PVP on the hydrolysis degree of –Si(OCH3)3 attached to ProTMS and MPTMS onto MNPs Fe3O4/PVP and Fe3O4 could be further monitored by TGA. From the TG and DTG curves (Fig. 3), the thermal decomposition of organic moieties in Fe3O4/PVP@SiO2/ProTMS occurred in two distinct steps: the first one in the temperature 200–500 °C range and the second one in the 500–600 °C range, accompanied by a continuous endothermic peak (see ESI†). It is noteworthy that Fe3O4/PVP@SiO2/ProTMS exhibited the 15.2% higher weight loss than Fe3O4@SiO2/ProTMS in the temperature range of 200–600 °C, although Fe3O4/PVP@SiO2/ProTMS contained a lower amount of Jørgensen–Hayashi catalyst 5. Another phenomenon was that a sharp weight loss of Fe3O4/PVP@SiO2/ProTMS (30.01%) was newly emerged in the 500–600 °C range (Fig. 3c). Based on the two points mentioned above, it was demonstrated that the hydrolysis efficiency of –Si(OCH3)3 decreased upon the attachment of ProTMS and MPTMS onto Fe3O4/PVP, and resulted in the increased SiOCH3 moiety due to the package of PVP, which was in accord with the stronger C–O stretching vibration at 1134 cm−1 in IR spectra of Fe3O4/PVP@SiO2/ProTMS and verified by the weakened C–O stretching vibration of 10th-recycled Fe3O4/PVP@SiO2/ProTMS. Furthermore, besides the same typical peaks of Fe3O4 at 30.3°, 35.6°, 43.3°, 53.8°, 57.4° and 63.0° in the XRD pattern, Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS also displayed the difference in the typical peak of SiO2 in the 16–25° range (Fig. 4a and b), which was closely evidenced to the hydrolysis degree of –Si(OCH3)3.
 | ||
| Fig. 3 Thermogravimetric curves of Fe3O4@SiO2/ProTMS (a), 10th-recycled Fe3O4/PVP@SiO2/ProTMS (b) and Fe3O4/PVP@SiO2/ProTMS (c). | ||
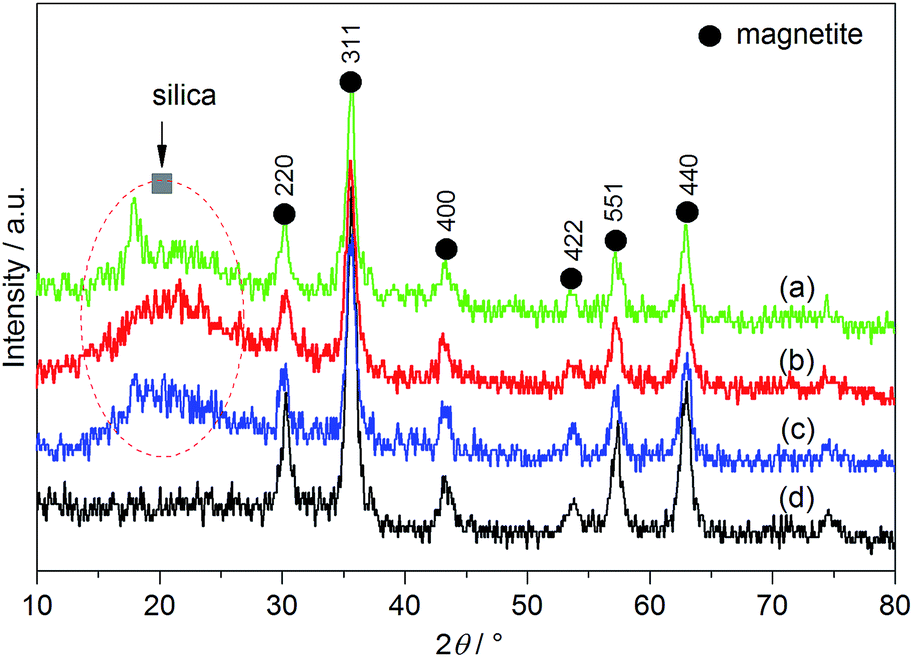 | ||
| Fig. 4 Powder X-ray diffraction patterns of Fe3O4/PVP@SiO2/ProTMS (a), Fe3O4@SiO2/ProTMS (b), 10th-recycled Fe3O4/PVP@SiO2/ProTMS (c) and Fe3O4 MNPs (d). | ||
The presence of PVP not only altered the chemical composition but also affected the morphology of MNPs-supported catalyst. The grafting of Jørgensen–Hayashi catalyst 5 onto MNPs Fe3O4 and Fe3O4/PVP led to a serious agglomeration due to the coated SiO2 generated from the hydrolysis of –Si(OCH3)3 moiety. However, seen from the SEM images, Fe3O4/PVP@SiO2/ProTMS exhibited the smaller even-textured and granular particles with the diameters ranging from 0.2 to 0.3 μm (Fig. 5f), compared with irregular Fe3O4@SiO2/ProTMS (Fig. 5b). The further investigation by the TEM image showed the densely agminated Fe3O4@SiO2/ProTMS (Fig. 5d) and nano-structured Fe3O4/PVP@SiO2/ProTMS (Fig. 5h) in an accumulative state upon being coated with SiO2 shell, respectively due to the profound and weakened hydrolysis of –Si(OCH3)3 in the absence or presence of PVP.
 | ||
| Fig. 5 The surface morphologies for TEM of MNPs Fe3O4 (a), SEM of Fe3O4@SiO2/ProTMS (b), TEM of Fe3O4@SiO2/ProTMS (c and d), TEM of MNPs Fe3O4/PVP (e), SEM of Fe3O4/PVP@SiO2/ProTMS (f), TEM of Fe3O4/PVP@SiO2/ProTMS (g and h). | ||
The nitrogen adsorption–desorption isotherm plots of all samples, obtained at 77 K, were shown in ESI.† After being stirred at room temperature polyvinylpyrrolidone (PVP) solution (10.0 mL, 25 g L−1) for 12 h, it was found that MNPs Fe3O4/PVP possessed the higher BET-specific surface area (65.8 m2 g−1), average pore diameter (11.7 nm) and pore volume (0.39 cm3 g−1) than bare Fe3O4 (7.9 m2 g−1, 5.2 nm and 0.021 cm3 g−1). Especially, Fe3O4/PVP had five characteristic cavities centered at 0.9, 1.4, 2.1, 3.4 and 6.6 nm, accompanied by some additional pores in the different sizes with the nitrogen desorption (Dv) (<1.0 × 10−3 mL Å−1 g−1) (Fig. 6c), whereas bare MNPs Fe3O4 displayed a broad pore size distribution (Fig. 6a). Interestingly, except for these five characteristic cavities, all the other pores in Fe3O4/PVP disappeared upon the immobilization of ProTMS and MPTMS through the hydrolysis of –Si(OCH3)3, which indicated that Jørgensen–Hayashi catalyst 5 was entrapped inside these disappeared pores. To our delight, the remained four characteristic pores of Fe3O4/PVP@SiO2/ProTMS with the high Dv values (>1.0 × 10−3 mL Å−1 g−1) provided the crucial channels for reactants to access catalytic sites in catalytic process. Unfortunately, Fe3O4@SiO2/ProTMS exhibited the non-uniform pore size distribution in the 1–10 nm range without the modification of PVP.
 | ||
| Fig. 6 Pore size distributions (PSDs) of Fe3O4 (a), Fe3O4@SiO2/ProTMS (b), Fe3O4/PVP (c) and Fe3O4/PVP@SiO2/ProTMS (d). | ||
Asymmetric Michael addition
To evaluate influence of PVP on the catalytic performance and reusability of Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS, the asymmetric Michael addition of propanal to trans-β-nitrostyrene was selected as a model reaction.In the Michael addition of propanal to trans-β-nitrostyrene, the corresponding syn-adducts, whose absolute stereochemistry was determined to be 2R,3S-configurations by comparing its optical rotation and HPLC spectra with literature values,18 were obtained as major stereoisomers in the high yields and excellent enantioselectivities (Table 1). To the best of our knowledge, it was found that Fe3O4/PVP@SiO2/ProTMS appeared as the most efficient MNPs-supported Jørgensen–Hayashi catalyst to date in terms of catalytic activity, stereoselectivity and reusability among the ever-reported Michael additions.11–13 Meanwhile, Fe3O4/PVP@SiO2/ProTMS (99%, syn/anti = 94/6, 98% ee) presented the superior catalytic performances to Fe3O4@SiO2/ProTMS (89%, syn/anti = 93/7 and 97% ee) (entry 18), which might be rationalized in terms of catalytic accessibility and steric confinement owing the above-mentioned structural texture and morphology resulted from the vital function of PVP. In the following catalytic reactions, Fe3O4/PVP@SiO2/ProTMS was chosen for the further optimization of experimental conditions.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mg)/catalyst (mol%) | Additive | Solvent | T (°C)/time (h) | Yieldb (%) | Syn/antic | eed (%) |
| a Reaction conditions: trans-β-nitrostyrene (40 mg, 0.27 mmol), propanal (94 mg, 1.62 mmol), additive (10 mol%), solvent (2.0 mL).b Isolated yield.c Determined by 1H NMR.d Determined by chiral HPLC.e 20 mol% of additive.f Fe3O4@SiO2/ProTMS used. | |||||||
| 1 | 30/7 | — | CHCl3 | 0/17 | 99 | 89/11 | 97 |
| 2 | 30/7 | TFA | CHCl3 | 0/8 | Trace | n.d. | n.d. |
| 3 | 30/7 | PhCO2H | CHCl3 | 0/12 | 93 | 92/8 | 97 |
| 4 | 30/7 | 2-FPhCO2H | CHCl3 | 0/12 | 95 | 90/10 | 97 |
| 5 | 30/7 | 3-NO2PhCO2H | CHCl3 | 0/8 | 99 | 96/4 | 98 |
| 6 | 30/7 | 3-NO2PhCO2He | CHCl3 | 0/24 | 93 | 93/7 | 97 |
| 7 | 30/7 | 3-NO2PhCO2H | C6H5CH3 | 0/8 | 87 | 88/12 | 95 |
| 8 | 30/7 | 3-NO2PhCO2H | CH2Cl2 | 0/8 | 94 | 90/10 | 96 |
| 9 | 30/7 | 3-NO2PhCO2H | EtOH | 0/8 | Trace | n.d. | n.d. |
| 10 | 60/14 | — | CHCl3 | 0/8 | 98 | 91/9 | 97 |
| 11 | 60/14 | 3-NO2PhCO2H | CHCl3 | 0/4 | 98 | 91/9 | 98 |
| 12 | 15/3.5 | — | CHCl3 | 0/32 | 76 | 92/8 | 97 |
| 13 | 15/3.5 | 3-NO2PhCO2H | CHCl3 | 0/22 | 96 | 89/11 | 98 |
| 14 | 30/7 | 3-NO2PhCO2H | CHCl3 | 20/5 | 99 | 87/13 | 96 |
| 15 | 30/7 | 3-NO2PhCO2H | CHCl3 | 10/7 | 99 | 90/10 | 97 |
| 16 | 30/7 | 3-NO2PhCO2H | CHCl3 | −10/20 | 86 | 96/4 | 98 |
| 17 | 30/7 | 3-NO2PhCO2H | CHCl3 | −20/32 | 82 | 98/2 | 98 |
| 18f | 26/7 | 3-NO2PhCO2H | CHCl3 | 0/8 | 89 | 93/7 | 97 |

Firstly, it was found that the acid strengths of Brønsted acids such as TFA (pKa = 0.23), 3-NO2C6H4CO2H (pKa = 2.45), 2-FC6H4CO2H (pKa = 3.27) and C6H4CO2H (pKa = 4.21) had a great effect on catalytic performances (entries 2–6). Disappointedly, TFA with the stronger acid strength only afforded a trace of Michael adduct (entry 2). Fortunately, the other weak Brønsted acids could effectively improve catalytic rate and stereoselectivity. Among them, 3-NO2C6H4CO2H (10 mol%) was proved to be the best effective co-catalyst with the excellent performances (99%, syn/anti = 96/4, 98% ee, entry 5). It was worthwhile to note that Fe3O4/PVP@SiO2/ProTMS gave the less catalytic performances (93%, syn/anti = 93/7, 97% ee, entry 6) after 24 h upon doubling the dosage of 3-NO2C6H5CO2H. Next, the effects of temperature, solvent and used amount of catalyst on catalytic performance were further screened out in detail. Our attempts to further reduce catalyst loading led to the decreased yield and stereoselectivity (entries 12 and 13), and ethanol as a solvent resulted in no activity (entry 9). Furthermore, although the decrease in temperature to −20 °C led to the product with the higher stereoselectivity (syn/anti = 98/2, 98% ee, entry 17), it took place at a considerably lower rate, requiring up to 32 h for the achievement of 82% yield.
The optimized protocol was expanded to a wide variety of trans-β-nitrostyrenes bearing electron-donating (–CH3, OCH3) and electron-withdrawing (–X) substituents at the o, m, p-positions. From Table 2, all trans-β-nitrostyrenes bearing electron-donating and electron-withdrawing substituents afforded the excellent catalytic performances at the same level (93–99%, syn/anti = 86–92:14–8, 96–98% ee, entries 1–9). It was worthwhile to note that the nitrostyrenes with o, p-OCH3 and o-CH3 electron-donating (entries 5, 7 and 9) and o-Cl electron-with-drawing substituent (entry 4) required respective 48 h and 24 h to achieve the 93–99% yields resulted from the electron-donating effect and vicinal hindrance of the substituents. A nitroalkene bearing a heterocyclic 2-furyl substituent also afforded a high 98% yield and 95% ee but gave a moderate diastereoselectivity (syn/anti = 81/19) (entry 10). Additionally, the Michael addition of propanal to aliphatic nitroalkene gave the good catalytic performances (97%, 97% ee, 86:14 dr) but needed a prolonged reaction period (34 h, entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | t (h) | Product | Yieldb (%) | Syn/antic | % eed |
| a Reaction conditions: nitrolefin (40 mg, 0.27 mmol), propanal (94 mg, 1.62 mmol), 3-NO2C6H4CO2H (10 mol%), CHCl3 (2.0 mL), 0 °C.b Isolated yield.c Determined by 1H NMR.d Determined by chiral HPLC. | |||||
| 1 | 8 |  |
98 | 88/12 | 97 |
| 2 | 7 |  |
99 | 87/13 | 97 |
| 3 | 9 |  |
95 | 86/14 | 96 |
| 4 | 24 |  |
98 | 88/12 | 96 |
| 5 | 48 |  |
99 | 92/8 | 97 |
| 6 | 12 |  |
97 | 89/11 | 96 |
| 7 | 48 |  |
93 | 92/8 | 96 |
| 8 | 12 |  |
98 | 89/11 | 96 |
| 9 | 48 |  |
95 | 90/10 | 97 |
| 10 | 11 |  |
98 | 81/19 | 95 |
| 11 | 34 | 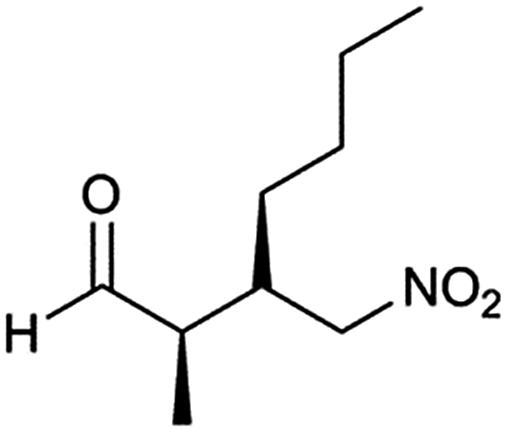 |
97 | 86/14 | 97 |
| 12 | 8 |  |
>99 | 96/4 | 98 |

Reusability of MNPs-supported catalyst
One of the main advantages associated with the supporting of a catalyst onto MNPs is the facile separation of a catalyst from reaction mixture with an external magnet. When the reaction was completed, an external magnet was put nearby the reaction flask to concentrate Fe3O4/PVP@SiO2/ProTMS, and the solution could be easily decanted. The recovered catalyst was directly reused for the next run after being washed with CHCl3 (2.0 mL × 3).Fig. 7 showed the comparative reusabilities of Fe3O4/PVP@SiO2/ProTMS and Fe3O4@SiO2/ProTMS including the yields and stereoselectivities. From Fig. 6, Fe3O4/PVP@SiO2/ProTMS displayed a better reusability than Fe3O4@SiO2/ProTMS. Unfortunately, the decreasing yields and diastereoselectivity catalyzed by Fe3O4@SiO2/ProTMS after the second run like the ever-reported Jørgensen–Hayashi catalysts using MNPs as a support.11–13 On the contrary, the yields catalyzed by Fe3O4/PVP@SiO2/ProTMS remained at the very high level (>94%) in the first five cycles. Even in 10th run, the 77% yield could be achieved after prolonging the reaction time to 48 h. More excitedly, the constant excellent stereoselectivity (98% ee and 96/4 dr) could be maintained in the ten consecutive cycles. To the best of our knowledge, Fe3O4/PVP@SiO2/ProTMS exhibited the most effective reusability among the reported MNPs-supported Jørgensen–Hayashi catalysts.
 | ||
| Fig. 7 The comparative reusability of Fe3O4@SiO2/ProTMS and Fe3O4/PVP@SiO2/ProTMS under the optimized reaction conditions. | ||
To understand the reason for the decreased yield, the structural change of the 10th-recycled Fe3O4/PVP@SiO2/ProTMS was monitored by TEM, TGA, elemental analysis and N2 adsorption–desorption isotherm. Above all, the increased carbon, hydrogen and nitrogen contents, especially the doubled nitrogen content (0.90% → 1.76%) stemmed from nitrogenous compounds such as nitroalkene, elucidated that the reactants and products were adsorbed on the porous backbone of the catalyst, which also verified by the decreased average pore size and pore volume from 15.7 nm and 0.031 cm3 g−1 to 2.5 nm and 0.021 cm3 g−1 respectively. Moreover, the structural change of SiO2 attached on the core of Fe3O4/PVP was elucidated by XRD and TGA, in which a wide and round XRD peak of SiO2 in the range of 16–25° (Fig. 3b) and a disappeared weight loss in the 500–600 °C range demonstrated the further hydrolysis of –SiOCH3. Meanwhile, it was found that the 10th-recycled Fe3O4/PVP@SiO2/ProTMS maintained its original magnetic property (Fig. 1b) and no agglomeration was observed by TEM (Fig. S10, ESI†). In conclusion, it was speculated that the adsorbed impurities and further hydrolysis of –SiOCH3 resulted in the structural changes such as pore volume and pore size distribution and was ultimately responsible for the decreased catalytic activity of Fe3O4/PVP@SiO2/ProTMS.
Conclusions
In summary, we had immobilized the homogeneous Jørgensen–Hayashi catalyst of (S)-diphenylprolinoltrimethylsilyl ether onto the backbone of Fe3O4/PVP via facile one-pot surface-modification, and successfully applied the MNPs Fe3O4-supported catalyst Fe3O4/PVP@SiO2/ProTMS in the asymmetric Michael addition of propanal to various nitroalkenes with excellent catalytic performances. Due to the adsorbed poly(vinylpyrrolidone) (PVP), Fe3O4/PVP@SiO2/ProTMS had a great change in chemical composition, surface morphology and pore structure related to the hydrolysis degree of –Si(OCH3)3, which provided a suitable micro-environment to achieve the good reusability with the high yields and unchangeable excellent stereoselectivities including syn/anti and % ee for the first time.Acknowledgements
We are grateful to the Fundamental Research Funds for the Central Universities (XDJK2015D028) and the National Science Foundation of China (621362005).Notes and references
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed; (b) J. Franzén, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjæsgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 18296–18304 CrossRef PubMed; (c) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821–3832 RSC.
- (a) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed; (b) A. Z. Halimehjani, I. N. N. Namboothiri and S. E. Hooshmand, RSC Adv., 2014, 4, 51794–51829 RSC; (c) H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS.
- (a) X. J. Li, B. L. Yang, X. B. Jia, M. Q. Chen and Z. G. Hu, RSC Adv., 2015, 5, 89149–89156 RSC; (b) S. Wang, P. W. Liu, W. J. Wang, Z. G. Zhang and B. G. Li, Catal. Sci. Technol., 2015, 7, 3798–3805 RSC; (c) C. Schafer, S. C. Mhadgut, N. Kugyela, M. Torok and B. Torok, Catal. Sci. Technol., 2015, 7, 716–723 RSC; (d) A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233–7241 CrossRef CAS PubMed; (e) K. Akagawa, S. Sakamoto and K. Kudo, Synlett, 2011, 6, 817–820 Search PubMed; (f) E. Alza and M. A. Pericàs, Adv. Synth. Catal., 2009, 351, 3051–3056 CrossRef CAS.
- (a) A. A. Elmekawy, J. B. Sweeney and D. R. Brown, Catal. Sci. Technol., 2015, 7, 716–723 Search PubMed; (b) C. L. Wu, X. Q. Long, X. K. Fu, G. W. Wang and Z. A. Mirza, RSC Adv., 2015, 5, 3168–3176 RSC; (c) M. Angeloni, O. Piermatti, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2014, 8, 1716–1726 CrossRef; (d) S. Calogero, D. Lanari, M. Orru, O. Piermatti, F. Pizzo and L. Vaccaro, J. Catal., 2011, 282, 112–119 CrossRef CAS; (e) W. Zheng, C. F. Lu, G. C. Yang, Z. X. Chen and J. Q. Nie, Catal. Commun., 2015, 62, 34–38 CrossRef CAS.
- (a) Y. Kong, R. Tan, L. L. Zhao and D. H. Yin, Green Chem., 2013, 15, 2422–2433 RSC; (b) X. Zhang, W. S. Zhao, C. K. Qu, L. L. Yang and Y. C. Cui, Tetrahedron: Asymmetry, 2012, 23, 468–473 CrossRef CAS.
- R. Tan, C. Y. Li, J. Q. Luo, Y. Kong, W. G. Zheng and D. H. Yin, J. Catal., 2013, 298, 138–147 CrossRef CAS.
- (a) R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Coord. Chem. Rev., 2015, 287, 137–156 CrossRef; (b) R. Dalpozzo, Green Chem., 2015, 17, 3671–3686 RSC; (c) J. W. Wan, L. ding, T. Wu, X. B. Ma and Q. Tang, RSC Adv., 2014, 4, 38323–38333 RSC.
- B. G. Wang, B. C. Ma, Q. Wang and W. Wang, Adv. Synth. Catal., 2010, 352, 2923–2928 CrossRef CAS.
- P. Riente, C. Mendoza and M. A. Pericás, J. Mater. Chem., 2011, 21, 7350–7355 RSC.
- M. Keller, A. Perrier, R. Linhardt, L. Travers, S. Wittmann, A. M. Caminade, J. P. Majoral, O. Reiser and A. Oualia, Adv. Synth. Catal., 2013, 355, 1748–1754 CrossRef CAS.
- A. B. Xia, C. Zhang, Y. P. Zhang, Y. J. Guo, X. L. Zhang, Z. B. Li and D. Q. Xu, Org. Biomol. Chem., 2015, 13, 9593–9599 CAS.
- (a) T. J. Yoon, J. S. Kim, B. G. Kim, K. N. Yu, M. H. Cho and J. K. Lee, Angew. Chem., Int. Ed., 2005, 44, 1068–1071 CrossRef CAS PubMed; (b) M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef CAS PubMed; (c) X. X. Zheng, S. Z. Luo, L. Zhang and J. P. Cheng, Green Chem., 2009, 11, 455–458 RSC.
- (a) A. Khalafi-Nezhad, E. S. Shahidzadeh, S. Sarikhani and F. Panahi, J. Mol. Catal. A: Chem., 2013, 379, 1–8 CrossRef CAS; (b) A. Khalafi-Nezhad and F. Panahi, Green Chem., 2011, 13, 2408–2415 RSC; (c) D. D. Feng, J. H. Xu, J. W. Wan, B. Xie and X. B. Ma, Catal. Sci. Technol., 2015, 5, 2141–2148 RSC.
- L. L. Lou, H. L. Du, Y. B. Shen, K. Yu, W. J. Yu, Q. Chen and S. X. Liu, Microporous Mesoporous Mater., 2014, 187, 94–99 CrossRef CAS; Y. Du, D. D. Feng, J. W. Wan and X. B. Ma, Appl. Catal., A, 2014, 479, 49–58 CrossRef; W. Wang, X. B. Ma, J. W. Wan, J. Cao and Q. Tang, Dalton Trans., 2012, 41, 5715–5726 RSC.
- (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed; (b) Z. L. Zheng, B. L. Perkins and B. Ni, J. Am. Chem. Soc., 2010, 132, 50–51 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, TEM and N2 adsorption–desorption isotherm of catalysts, 1H, 13C NMR and HPLC spectra of intermediates and products. See DOI: 10.1039/c6ra01051b |
| This journal is © The Royal Society of Chemistry 2016 |