
Significant improvement in the hydrogen storage capacity of a reduced graphene oxide/TiO2 nanocomposite by chemical bonding of Ti–O–C†
Zahra Gohari Bajestania,
Alp Yürümb and
Yuda Yürüm*a
aFaculty of Engineering and Natural Sciences, Sabanci University, Tuzla, Istanbul, 34956, Turkey. E-mail: yyurum@sabanciuniv.edu
bSabanci University Nanotechnology Research and Application Center, Tuzla, Istanbul, 34956, Turkey
First published on 29th March 2016
Abstract
A series of graphene-based nanocomposites with different TiO2 contents have been prepared via a facile chemical method. All nanocomposites were employed as hydrogen gas adsorption materials at room temperature and pressures up to 10 bar. The effect of dispersion state and size of the particles on the hydrogen storage capacity of nanocomposites was studied. The highest hydrogen uptake of 0.39 wt% was obtained among prepared nanocomposites and it is 125% higher than the hydrogen adsorption of the parent graphene material. This improvement can account for the presence of a high number of active sites needed for hydrogen molecules and the strong interaction between nanoparticles and graphene sheets.
1. Introduction
In recent years, production, storage, and consumption of energy have become a serious global problem. Hydrogen attracts considerable attention in the energy area since it can be a clean energy resource for the next-generation energy carriers in mobile and stationary power systems.1 However, finding materials for efficient hydrogen storage is one of the most important issues in future of renewable energies.2,3 Several candidates were studied during the last decades as safe and effective hydrogen adsorbents but none of them met the storage targets set by the U.S. Department of Energy (DOE).4–6 Among these options, graphene a single atomic layer sheet of sp2 bonded carbon atoms, has been cited as a promising candidate for energy conversion and storage applications due to its high theoretically surface area, large microporosity, and good chemical stability.7–10 Nevertheless, the range of reported hydrogen uptakes of graphene at ambient conditions is low.11 Srinivas et al.12 and Huang et al.4 reached hydrogen uptake of 0.1 wt% at 10 bar and 0.067 wt% at 57 bar, respectively. Thermodynamically, hydrogen molecules are incapable of penetrating between the graphene layers in graphitic structure whereby H2 adsorption generally restricts to the outermost sheets. Weak binding energy between carbon nanostructure and H2 at ambient conditions is also another key factor that leads to low H2 adsorption capacity of this material.8 Thus, surface modifications, such as doping and functionalization have been proposed to enhance gas adsorption property of graphene based materials.8,13 So far many theoretical and experimental studies have been devoted to functionalization of graphene by metals2,10,14,15 and metal oxides16–18 in which metal oxides offer a great scope for tailor-made carbonaceous adsorbents. Due to various advantages e.g., suppressing the re-stacking of graphene layers, good bonding and interfacial interactions with oxygen-containing groups on graphene sheets, fabrication of graphene/metal oxide is expected to be an effective and practical method toward increasing the hydrogen storage capacity of graphene.13,17TiO2 has been widely studied due to its photochemical, catalytic and dielectric characteristics. Theoretical studies showed the potential of Ti/TiO2-anchored carbonaceous materials17,19,20 as a hydrogen storage material for room-temperature applications. Recent experimental results on TiO2-decorated expanded graphite21 and TiO2-carbon nanotubes (CNT) composites22,23 revealed the enhanced electrochemical hydrogen uptake of graphene and higher H2 gas adsorption of CNT after impregnation with TiO2 nanoparticles.
In this work, TiO2-integrated graphene nanocomposites with different amounts of TiO2 nanoparticles were prepared via a facile chemical method. We focused on the hydrogen adsorption behavior of these nanocomposites at room temperature and low pressures that are relevant for practical on-board storage systems. Effects of the loading, dispersion state and size of nanoparticles on hydrogen storage capacity of nanocomposites were studied. Homogenous dispersion of TiO2 nanoparticles with diameter of <20 nm was found to enhance the hydrogen storage capacity of parent graphene by 125%. Higher hydrogen uptake of nanocomposites compared to that of graphene sample was related to strong attachment of highly distributed nanoparticles to the underlying graphene sheets.
2. Experimental section
2.1 Materials
Natural graphite flake (99%), sulfuric acid (H2SO4, 98%), sodium nitrate (NaNO3), potassium permanganate (KMnO4) and hydrochloric acid (HCl, 37%) were purchased from Sigma-Aldrich. Hydrogen peroxide (H2O2, 30%) and titanium chloride (TiCl3) were obtained from Merck. All reagents were analytical grade and used without further purification.2.2 Preparation
Graphite oxide was prepared from natural graphite flakes using Hummers' method.24 In order to prepare TiO2-integrated graphene sheets, hereafter referred to as RGO–T nanocomposites, graphite oxide was first sonicated (Vibra Cell 75041, Bioblock Scientific) in water for 2 h to obtain a homogenous dispersion of graphene oxide (GO) in aqueous medium. Different amounts of TiCl3 were then added to the mixture to reach Ti loading of 3 wt%, 5 wt%, and 7 wt%. After stirring for 24 h, suspensions were filtered, vacuum-dried at 60 °C and reduced at 1000 °C for 12 min under argon atmosphere to obtain reduced graphene oxide (RGO) with different amounts of TiO2, named as RGO–T3, RGO–T5, and RGO–T7 in accordance with increasing amount of TiCl3 in the mixing step. Thermogravimetric analysis indicated the presence of ∼10 wt%, ∼12 wt% and ∼15 wt% TiO2 in RGO–T3, RGO–T5, and RGO–T7, respectively (Fig. S1, ESI†). As a reference, GO (without addition of TiCl3) was thermally reduced at 1000 °C to attain fully exfoliated reduced graphene oxide.2.3 Characterization
X-Ray diffraction (XRD) patterns were performed using Bruker AXS diffractometer fitted with a Siemens X-ray gun using 0.15406 nm Cu Kα radiation. Raman spectroscopic analysis was carried out using Renishaw inVia reflex Raman spectrometer with a 532 nm laser beam in the range of 100–3500 cm−1 while samples were loaded on silicon wafer and focused with a 50× objectives. X-ray photoelectron spectroscopy (XPS) analyses were conducted on a Thermo K-alpha X-ray photoelectron spectrometer with a monochromated Al Kα supported by a low energy electron/ion flood gun for charge neutralization. Transmission electron microscopy (TEM) micrographs and scanning electron microscopy (SEM) images were taken by Jeol 2000FX with the accelerating voltage of 200 kV and Leo Supra 35VP field emission scanning electron microscope with an acceleration voltage of 2 kV, respectively. The Brunauer–Emmett–Teller (BET) specific surface area and porosity of samples were determined by analyzing the standard nitrogen adsorption isotherms at 77 K using nova 2200e, Quantachrome instruments. Hydrogen storage capacity of samples was measured by using Intelligent Gravimetric Analyzer (IGA 001, Hiden Isochema). All nanocomposites (20–25 mg) were degassed at 100 °C for 12 h under high vacuum (∼10−7 mbar) prior to measurements and then, hydrogen adsorption isotherms were measured at 298 K up to 10 bar.3. Results and discussion
3.1 Materials characterization
X-ray diffraction was first carried out to determine the crystallographic structure of the samples. As shown in Fig. 1a, natural graphite flakes show a sharp peak at 2θ = 26° that corresponds to (002) plane and an interlayer spacing (d-spacing) of 3.3 Å. Upon oxidation, the (002) peak disappeared and a low intensity peak, indexed as (001), emerged at 2θ ≈ 10.5° with d-spacing of 8.4 Å. This increase of the d-spacing after oxidation has been correlated with intercalation of oxygen-containing groups between graphene layers that leads to change of the crystallographic structure of graphite.4 After inclusion of Ti, intensity and full width at half maximum (FWHM) of the characteristic peak of graphite oxide (2θ ≈ 10.5°) were changed. In addition, a new peak centered at 2θ ≈ 25° was observed in GO–T5 and GO–T7 whereas no peak was detected at this region in GO–T3. Given the characteristic peak of TiO2 (101) at 2θ ≈ 25°,25 former observation can be attributed to formation of TiO2 in the samples. On the other hand, absence of this peak in GO–T3 can be related to the low metal content and/or small size of particles.4 Besides, progressive increase of FWHM and decrease of the intensity of GO main peak by Ti content suggests that GO was partially deoxygenated in the presence of Ti and reduced to turbostratic graphite.26 Consequently, appearance of the peak at 2θ ≈ 25° can also represent the formation of turbostratic graphite structures in the sample. Indeed, formation of turbostratic graphite coincides with the formation of TiO2 particle in specimens. According to Li et al.,27 increase of FWHM of graphene layers correlates with non-uniformity in d-spacing, curvature and more importantly distortion of the layers. As a result, Ti component which intercalates between graphene oxide layers possibly forms structural defects in the system. Fig. 1b shows XRD patterns of thermally reduced GO samples before and after addition of Ti. Observation of a broad and low-intensity (002) peak in the absence of (001) peak in RGO indicates the complete reduction of graphite oxide sheets and formation of poorly ordered graphene-like structure along the stacking direction. On the same basis, absence of GO peak at ≈10.5° was used as a clear indication of reduction of graphite oxide sheets in RGO–T nanocomposites.25,28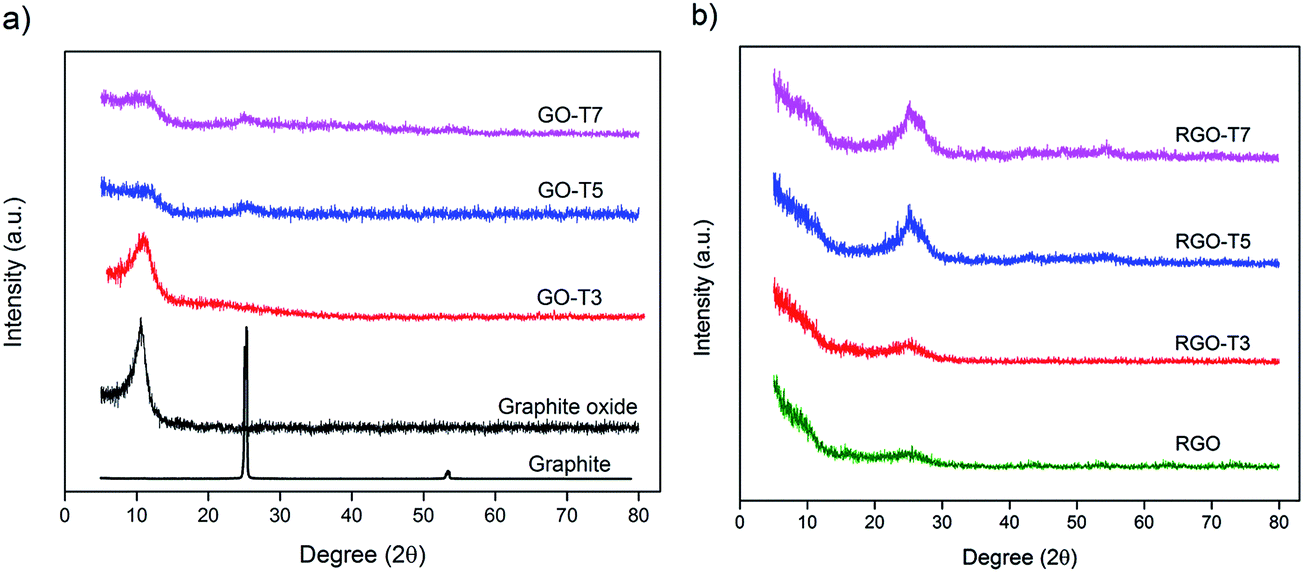 | ||
| Fig. 1 XRD patterns of (a) GO and (b) RGO samples with increasing amount of Ti addition. | ||
Raman spectroscopy was used to study the structural properties of particles and quality of graphene sheets, particularly defects and order in the system.24 Fig. 2 displays the Raman spectra of graphite oxide, RGO and RGO–T nanocomposites with typical characteristic bands at ∼1600 cm−1 and ∼1350 cm−1, known as G-band and D-band, respectively. G-band corresponds to vibration of the sp2-bonded carbon atoms in a 2D hexagonal lattice while D-band represents in-plane stretching motion of symmetric sp2 C–C bonds.29,30 To analyze the degree of disorder in graphitic layers, intensity ratio of the D and G bands (ID/IG) has been widely used in literature.3,29,30 The ID/IG value of 0.88 was calculated from the Raman spectrum of GO sample while higher ID/IG values were observed in the Raman spectra of RGO and RGO–T nanocomposites. This increased ID/IG ratio, points out the formation of additional defects during the deposition of particles that is a result of the interaction between particles and graphene sheets.3,31 Besides the G and D bands, a low intensity peak emerged at 151 ± 1 cm−1 in RGO–T nanocomposites. This peak was assigned to Eg mode of TiO2 and arises from the external vibration of anatase phase.32–35 Inset of Fig. 2 magnifies the wavenumber of 100–900 cm−1 in RGO–T7 and shows two vibration peaks at 400 cm−1 (B1g(1)) and 635 cm−1 (Eg(2)) in addition to Eg mode of TiO2. This observation confirms the formation of anatase TiO2 in RGO–T nanocomposites after thermal reduction process.32–35 However, a large frequency blue-shift of Eg was detected in RGO–T nanocomposites compared to Eg of anatase single crystal (144 cm−1). A similar large frequency shift was also reported by Zheng et al.36 for TiO2 nanocrystals fabricated by solution chemical process. They showed when dimensions of TiO2 crystallites decrease to nanometer scale, frequency shift occurs in Eg mode as a result of phonon confinement.
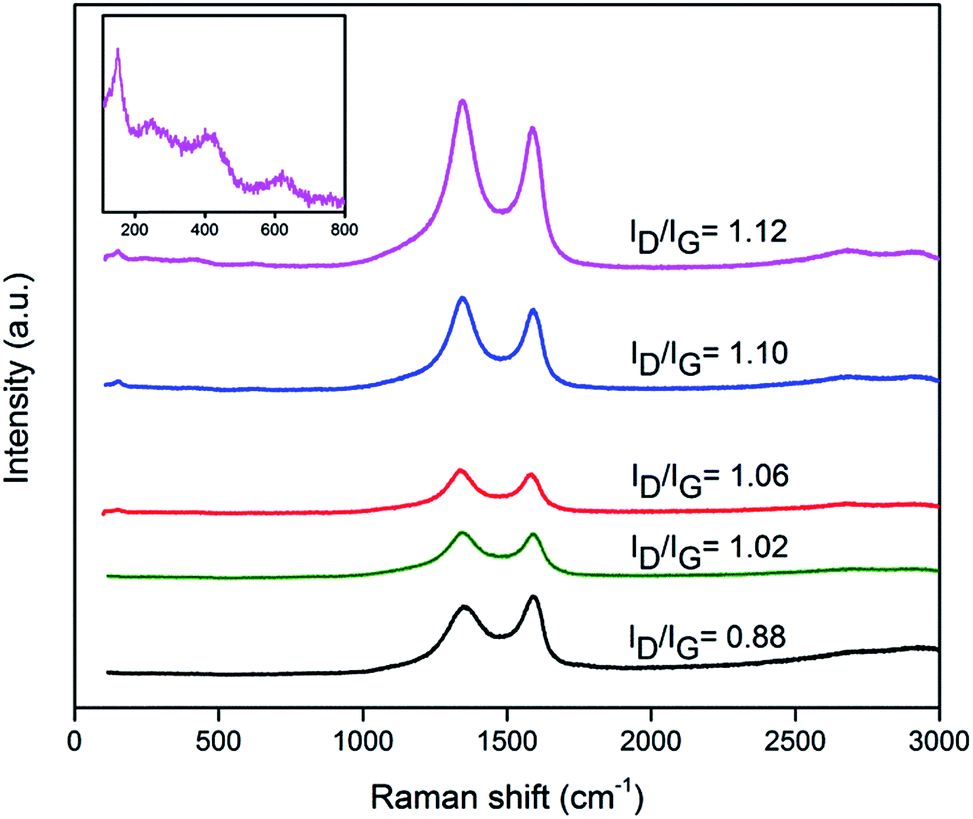 | ||
| Fig. 2 Raman spectra of graphite oxide, RGO and RGO–T nanocomposites, from bottom to top: graphite oxide, RGO, RGO–T3, RGO–T5 and RGO–T7. Inset magnifies the wavenumber of 100–900 cm−1 in RGO–T7. | ||
Distribution and size of the particles in RGO–T nanocomposites were studied using SEM, EDS and TEM. High magnification SEM images of samples (Fig. 3a–c) clearly demonstrate that the parent graphene sheets were coated with TiO2 nanoparticles. RGO–T3 and T5 display homogenous dispersion of nanoparticles while excess amount of Ti led to inhomogeneous deposition, growth and formation of submicron aggregation of nanoparticles in RGO–T7. The elemental analysis of RGO–T3 was provided in Fig. 3d–f as representative EDS results of RGO–T nanocomposites. Detection of distributed patterns of Ti and O in the elemental mapping confirms the homogeneous deposition of TiO2 nanoparticles throughout the sample.
 | ||
| Fig. 3 (a) SEM images of RGO–T3, (b) RGO–T5, and (c) RGO–T7, (d) SEM image of RGO–T3 with low magnification, (e) Ti and (f) O elemental mapping of same area shown in (d). | ||
TEM micrographs of RGO and RGO–T nanocomposites are presented in Fig. 4a–d. Wrinkled structured graphene sheets were clearly observed that indicates the formation of few layer graphene layers in all samples after thermal exfoliation. Homogeneous dispersion of TiO2 nanoparticles was detected as dark spots in TEM micrographs shown in Fig. 4b–d. Size of TiO2 nanoparticles in RGO–T3 was found to be 10 ± 5 nm whereas higher amounts of Ti addition introduced higher average of particle sizes in RGO–T7. According to Wu et al.17 and Wang et al.,37 size, morphology and anchoring of nanocrystals are dependent on the degree of oxidation of the underlying substrate. It is thought that Ti component could use the oxygen functional groups as nucleation centers. As a result, highly oxidized GO surface with high concentration of defects interacts strongly with particles and this strong pinning force hinders the diffusion and growth of formed particles.31,38 Tsao et al.39 showed that size of the catalyst particles plays an important role in H2 uptake in which Pt nanoparticles of 1–2 nm remarkably increased the H2 storage capacity of Pt/activated carbon nanocomposite. Therefore, formation of fine and distributed nanoparticles is highly preferred in order to increase the hydrogen adsorption capacity of RGO–T nanocomposites.
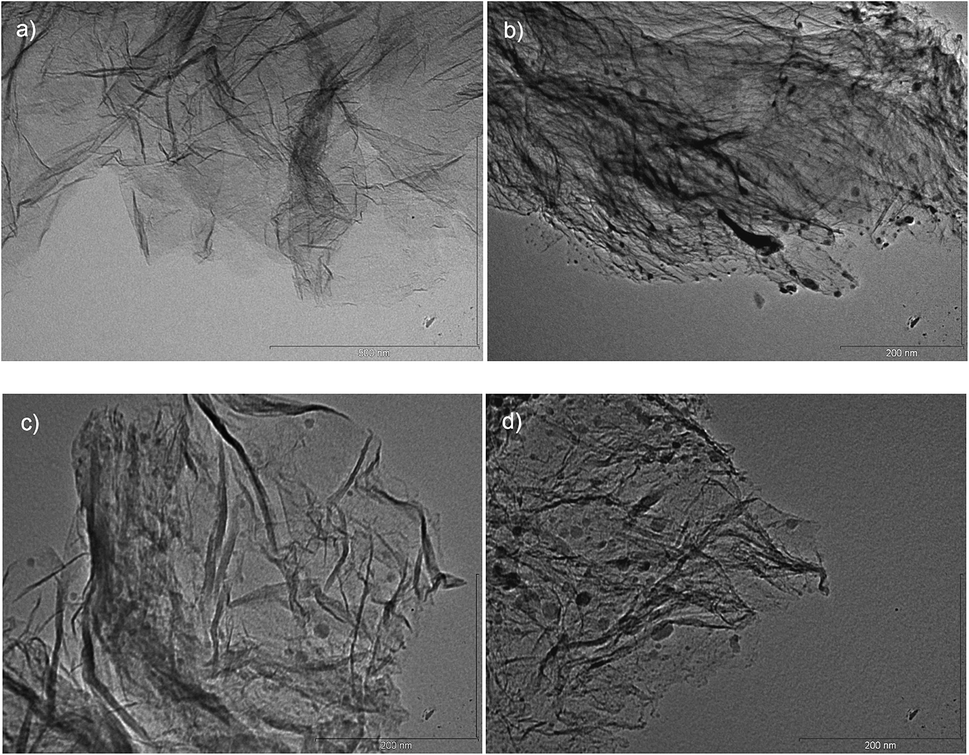 | ||
| Fig. 4 TEM micrograph of (a) RGO, (b) RGO–T3, (c) RGO–T5, and (d) RGO–T7. | ||
XPS measurement was performed to elucidate the chemical state of elements present in graphite oxide, RGO and RGO–T nanocomposites. Fig. 5a depicts a representative XPS survey spectrum (RGO–T5) that shows specimens mainly consist of C and O with no trace of contamination. The presence of Ti was detected in RGO–T nanocomposites with a weak signal at ∼459 eV that corresponds to Ti 2p electrons.40 Fig. 5b depicts C 1s XPS spectrum of graphite oxide with four peaks centered at 284.56, 285.02, 286.8 and 288.8 eV. These peaks were assigned to the C–C (aromatic),41 C–OH,31 C (epoxy/alkoxy)/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O42 and OC–O (carboxylic)35 groups respectively that imply the highly oxidized state of graphene sheets. After thermal reduction, the intensity of components associated with oxygenated functional groups significantly decreased (Fig. 5c). This decrease illustrates that most of the oxygen-containing functional groups are removed and GO is transformed to graphene.35 In Fig. 5d, Ti 2p XPS spectra of RGO–T nanocomposites exhibit two peaks centered at 464.7 ± 0.1 and 459 ± 0.2 eV assigned respectively to the Ti 2p1 and Ti 2p3 spin–orbital splitting photoelectrons in the Ti4+ state. The splitting energy between two Ti-bands was 5.69 ± 0.02 eV that is in agreement with the normal state of Ti4+.25,43,44 As TiO2 content was increased, Ti 2p3 peak narrowed (decrease of FWHM) and binding energies of Ti 2p feature shifted toward higher binding energies. According to Oh et al.,45 increasing thickness of deposited TiO2 shifts the bonding energy of Ti core level to higher values and decreases the FWHM of Ti 2p3 peak. Therefore, observation of shifting and narrowing Ti 2p3 peak with Ti content can be related to change of the dimensions of nanoparticles that is in agreement with TEM micrographs of RGO–T nanocomposites.
O42 and OC–O (carboxylic)35 groups respectively that imply the highly oxidized state of graphene sheets. After thermal reduction, the intensity of components associated with oxygenated functional groups significantly decreased (Fig. 5c). This decrease illustrates that most of the oxygen-containing functional groups are removed and GO is transformed to graphene.35 In Fig. 5d, Ti 2p XPS spectra of RGO–T nanocomposites exhibit two peaks centered at 464.7 ± 0.1 and 459 ± 0.2 eV assigned respectively to the Ti 2p1 and Ti 2p3 spin–orbital splitting photoelectrons in the Ti4+ state. The splitting energy between two Ti-bands was 5.69 ± 0.02 eV that is in agreement with the normal state of Ti4+.25,43,44 As TiO2 content was increased, Ti 2p3 peak narrowed (decrease of FWHM) and binding energies of Ti 2p feature shifted toward higher binding energies. According to Oh et al.,45 increasing thickness of deposited TiO2 shifts the bonding energy of Ti core level to higher values and decreases the FWHM of Ti 2p3 peak. Therefore, observation of shifting and narrowing Ti 2p3 peak with Ti content can be related to change of the dimensions of nanoparticles that is in agreement with TEM micrographs of RGO–T nanocomposites.
 | ||
| Fig. 5 (a) XPS survey spectra, (b) C 1s XPS spectrum of graphite oxide, (c) C 1s XPS spectrum of RGO, (d) Ti 2p XPS spectra of RGO–T, (e) C 1s XPS spectra of RGO–T nanocomposite, and (f) O 1s XPS spectra of graphite oxide and RGO–T nanocomposites. | ||
C 1s XPS spectra of RGO–T nanocomposites are presented in Fig. 5e with superimposition of C 1s XPS of RGO for comparison. All RGO–T nanocomposites show the main peak at 284.7 ± 0.2 eV (C–C aromatic band) in which the FWHM of the peak increased from 1.02 eV in RGO to 1.3, 2.01 and 1.9 eV in RGO–T3, T5 and T7, respectively. The FWHM of the C 1s core level band has been used to evaluate the degree of chemical and structural heterogeneity in environment of carbon atoms.46,47 Therefore, it can be concluded that environment of carbon atoms becomes more heterogeneous through the addition of Ti suggesting the bonding of TiO2 nanoparticles with C atoms of graphene sheets.
To further support the presence of this bonding, O 1s XPS spectra of graphite oxide and RGO–T nanocomposites are depicted in Fig. 5f. The O 1s spectrum of graphite oxide shows a peak centered at 532.5 eV that is closely related to the hydroxyl groups on the surface of graphite oxide sheets. However, this peak shifted to 530.6 ± 0.1 eV in RGO–T nanocomposites. In agreement with previous reports in bonding of Ti atoms with any available oxygen to form TiO2,48,49 this peak was assigned as bonding energy of O in Ti–O–C bond.50,51 As a result, bonding between Ti, O, and C confirms the integration of nanoparticles into graphene sheets.
3.2 Gas adsorption behavior
Nitrogen adsorption–desorption isotherm was employed to characterize the specific surface area and pore structure of nanocomposites (Fig. 6a). According to the IUPAC classification, all samples showed a nature of type IV curve, that is, a low N2 adsorption capacity at low relative pressure (P/P0) followed by a hysteresis loop at high P/P0. The former indicates the presence of small number of micropores while the latter reflects the formation of mesoporous structures in the system.52 Higher N2 adsorption of pristine graphene than that of RGO–T illustrates the lower adsorption capability of RGO after deposition of nanoparticles when physical adsorption is the dominant process.2 | ||
| Fig. 6 (a) N2 adsorption isotherms and (b) pore size distribution of RGO and RGO–T nanocomposites. | ||
The Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halenda (BJH) methods were applied to determine the specific surface area and pore structure, respectively. The pore-size distribution isotherm of RGO (Fig. 6b) displayed one peak concentrated in 2–5 nm. After incorporation of nanoparticles, the main peak was weakened and an extra peak emerged at lower radius (∼1 nm). Moreover, significant decrease in average pore size was detected after addition of Ti to the samples (Table 1). As a result, the reduction in specific surface area and average pore size can be attributed to the partial blockage of mesopores by TiO2 nanoparticles.53
| Specific surface area (m2 g−1) | Pore diameter (Å) | |
|---|---|---|
| RGO | 461.76 | 25.5 |
| RGO–T3 | 371.31 | 8.1 |
| RGO–T5 | 219.35 | 8.3 |
| RGO–T7 | 207.50 | 7.8 |
Fig. 7 presents the hydrogen adsorption isotherms of RGO and RGO–T nanocomposites at 298 K. As expected, the hydrogen uptake of all samples increased by hydrogen pressure. The parent graphene sample showed hydrogen uptake of 0.17 wt% at 10 bar. After TiO2-integration, the hydrogen storage capacity of 0.39 wt% was obtained by RGO–T5 that is 125% higher than that of pristine graphene. Further addition of Ti (RGO–T7) caused a decrease in hydrogen adsorption, possibly owing to aggregation of nanoparticles and losing porosity/surface area.
 | ||
| Fig. 7 Hydrogen adsorption isotherms RGO and RGO–T nanocomposites. | ||
The enhanced hydrogen storage capacity of the nanocomposites cannot be attributed to differences between the surface areas of samples since all nanocomposites displayed lower BET specific surface area than RGO (Fig. 6a). In addition, comparison of hydrogen adsorption isotherms at 298 K reveals the change of the shape of the adsorption curves from concave in pristine graphene to relatively linear in RGO–T samples. Li et al.54 found that linear adsorption isotherm is the characteristic behavior of hydrogen spillover on nearly all adsorbents and is very different from that of physical adsorptions characterized by an isotherm concave to the pressure axis. Thus, the higher hydrogen adsorption capacity of RGO–T nanocomposites originates from a process other than physical adsorption. Wang et al.19,55 have shown by the first-principle computations that strongly anchored Ti atoms on surface of GO template are superior sites for hydrogen adsorption whereby each Ti atom can bind multiple H2 with the binding energies of 14–41 kJ mol−1 H2. Results of hydrogen storage measurement obtained by Mishra et al.,56 Zhang et al.,22 and Lueking and Yang57 suggest the contribution of chemical adsorption in hydrogen uptake of different carbonaceous materials after incorporation of TiO2. They showed that this contribution can be attributed to non-classical s–p–d hybridization.22 As a result, higher hydrogen storage capacity of RGO–T nanocomposites compared to pristine graphene can be linked to the activation of processes usually grouped as chemisorption. Lueking and Yang57 have also underscored the role of catalyst-support interaction for improvement of hydrogen uptake of composites in which simple mixing of metal particles with support does not improve the hydrogen uptake of the system. Besides, Choucair and Mauron58 reported decrease in hydrogen uptake of graphene at 77 K and 1 atm H2 pressure after mechanical mixing with 1/1 weight ratio TiO2 nanoparticles. Given the importance of particle size39 and dispersion of nanoparticles,59 strong attachment of highly distributed nanoparticles to the underlying graphene sheets was found to be essential to improve the hydrogen uptake of graphene/metal oxide nanocomposites.
4. Conclusions
In this work, a series of TiO2-integrated reduced graphene oxide composites with different amounts of TiO2 were prepared via a facile chemical impregnation method. By observation of TEM micrographs, TiO2 nanoparticles with diameter of less than 20 nm were homogeneously dispersed on the graphene sheets. The effect of metal oxide content on hydrogen adsorption of nanocomposites at ambient temperature and pressures up to 10 bar was studied. It was shown that strong interaction of metal oxide nanoparticles with the support, smaller particle size, and high dispersion of nanoparticles are necessary to promote the hydrogen storage capacity of nanocomposites. The highest storage capacity of 0.39 wt% was obtained among all nanocomposites and it is 125% higher than that of parent graphene material. This significant improvement of hydrogen adsorption compared to RGO–TiO2 obtained by mechanical mixing (reported by ref. 58) was correlated with the formation of Ti–O–C bonding between nanoparticles and graphene substrate. We believe that the use of oxygen functional groups of graphene oxide sheets to form this bonding can also be extended to other types of metal oxide/carbonaceous nanocomposites.Acknowledgements
Zahra Gohari would like to thank PhD student Omid Akhlaghi for SEM and EDS characterizations.References
- K. Spyrou, D. Gournis and P. Rudolf, ECS J. Solid State Sci. Technol., 2013, 2, M3160–M3169 CrossRef CAS
.
- C.-H. Chen, T.-Y. Chung, C.-C. Shen, M.-S. Yu, C.-S. Tsao, G.-N. Shi, C.-C. Huang, M.-D. Ger and W.-L. Lee, Int. J. Hydrogen Energy, 2013, 38, 3681–3688 CrossRef CAS
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem.–Eur. J., 2011, 17, 10763–10770 CrossRef CAS PubMed
- C.-C. Huang, N.-W. Pu, C.-A. Wang, J.-C. Huang, Y. Sung and M.-D. Ger, Sep. Purif. Technol., 2011, 82, 210–215 CrossRef CAS
- H.-P. Zhang, X.-G. Luo, X.-Y. Lin, X. Lu and Y. Leng, Int. J. Hydrogen Energy, 2013, 38, 14269–14275 CrossRef CAS
- B. P. Vinayan, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2013, 38, 2240–2250 CrossRef CAS
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS
- S. Gadipelli and Z. X. Guo, Prog. Mater. Sci., 2015, 69, 1–60 CrossRef CAS
- W. G. Hong, B. H. Kim, S. M. Lee, H. Y. Yu, Y. J. Yun, Y. Jun, J. B. Lee and H. J. Kim, Int. J. Hydrogen Energy, 2012, 37, 7594–7599 CrossRef CAS
- L. Ma, J.-M. Zhang and K.-W. Xu, Appl. Surf. Sci., 2014, 292, 921–927 CrossRef CAS
- A. G. Klechikov, G. Mercier, P. Merino, S. Blanco, C. Merino and A. V. Talyzin, Microporous Mesoporous Mater., 2015, 210, 46–51 CrossRef CAS
- G. Srinivas, Y. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Carbon, 2010, 48, 630–635 CrossRef CAS
- V. Tozzini and V. Pellegrini, Phys. Chem. Chem. Phys., 2013, 15, 80–89 RSC
- A. Lebon, J. Carrete, L. J. Gallego and A. Vega, Int. J. Hydrogen Energy, 2015, 40, 4960–4968 CrossRef CAS
- J. Lu, Y. Guo, Y. Zhang, Y. Tang and J. Cao, J. Solid State Chem., 2015, 231, 53–57 CrossRef CAS
- Z. Wang and C.-J. Liu, Nano Energy, 2015, 11, 277–293 CrossRef CAS
- Z.-S. Wu, G. Zhou, L.-C. Yin, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS
- S. Yang, Y. Sun, L. Chen, Y. Hernandez, X. Feng and K. Müllen, Sci. Rep., 2012, 2, 427 Search PubMed
- L. Wang, K. Lee, Y.-Y. Sun, M. Lucking, Z. Chen, J. J. Zhao and S. B. Zhang, ACS Nano, 2009, 3, 2995–3000 CrossRef CAS PubMed
- E. Liu, Y. Gao, N. Zhao, J. Li, C. He and C. Shi, J. Appl. Phys., 2013, 113, 153708 CrossRef
- Y. Yu, N. Zhao, C. Shi, C. He, E. Liu and J. Li, Int. J. Hydrogen Energy, 2012, 37, 5762–5768 CrossRef CAS
- Z. Zhang, J.-Y. Hwang, M. Ning and X. Li, Int. J. Hydrogen Energy, 2012, 37, 16018–16024 CrossRef CAS
- S.-U. Rather, N. Mehraj-ud-din, R. Zacharia, S. W. Hwang, A. R. Kim and K. S. Nahm, Int. J. Hydrogen Energy, 2009, 34, 961–966 CrossRef CAS
- H. L. Poh, F. Sanek, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515–3522 RSC
- M. S. A. Sher Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 CAS
- S. Li, Z. Chen, Y. Jin, S. Chen, H. Wang, J. Geng, Q. Song, X. Yang, L. Ma, S. Li, Z. Qin and C. Zheng, Solid State Sci., 2011, 13, 862–866 CrossRef CAS
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS
- A. Dhanabalan, X. Li, R. Agrawal, C. Chen and C. Wang, Nanomaterials, 2013, 3, 606 CrossRef CAS
- M. Giovanni, H. L. Poh, A. Ambrosi, G. Zhao, Z. Sofer, F. Sanek, B. Khezri, R. D. Webster and M. Pumera, Nanoscale, 2012, 4, 5002–5008 RSC
- X. Wang and X. Zhang, Electrochim. Acta, 2013, 112, 774–782 CrossRef CAS
- G. Compagnini, P. Russo, F. Tomarchio, O. Puglisi, L. D'Urso and S. Scalese, Nanotechnology, 2012, 23, 505601 CrossRef CAS PubMed
- C. Chang-Jun, X. Mao-Wen, B. Shu-Juan, J. Chen-Chen, L. Zhen-Jiang and J. Dian-Zeng, Nanotechnology, 2013, 24, 275602 CrossRef PubMed
- H. C. Choi, Y. M. Jung and S. B. Kim, Vib. Spectrosc., 2005, 37, 33–38 CrossRef CAS
- H. Yoshitake and D. Abe, Microporous Mesoporous Mater., 2009, 119, 267–275 CrossRef CAS
- K. Li, T. Chen, L. Yan, Y. Dai, Z. Huang, J. Xiong, D. Song, Y. Lv and Z. Zeng, Colloids Surf., A, 2013, 422, 90–99 CrossRef CAS
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912 CrossRef CAS
- H. Wang, J. T. Robinson, G. Diankov and H. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS PubMed
- C. Xu and X. Wang, Small, 2009, 5, 2212–2217 CrossRef CAS PubMed
- C.-S. Tsao, Y.-R. Tzeng, M.-S. Yu, C.-Y. Wang, H.-H. Tseng, T.-Y. Chung, H.-C. Wu, T. Yamamoto, K. Kaneko and S.-H. Chen, J. Phys. Chem. Lett., 2010, 1, 1060–1063 CrossRef CAS
- I. Iatsunskyi, M. Kempiński, G. Nowaczyk, M. Jancelewicz, M. Pavlenko, K. Załęski and S. Jurga, Appl. Surf. Sci., 2015, 347, 777–783 CrossRef CAS
- L. Yue-Wen, G. Meng-Xue, F. Lan, D. Shun-Liu, B. Jian-Feng, X. Su-Yuan, C. Zhong, H. Rong-Bin and Z. Lan-Sun, Nanotechnology, 2013, 24, 025604 CrossRef PubMed
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640–1645 RSC
- X. Bai, X. Zhang, Z. Hua, W. Ma, Z. Dai, X. Huang and H. Gu, J. Alloys Compd., 2014, 599, 10–18 CrossRef CAS
- S. Bourgeois, P. le Seigneur and M. Perdereau, Surf. Sci., 1995, 328, 105–110 CrossRef CAS
- W. S. Oh, C. Xu, D. Y. Kim and D. W. Goodman, J. Vac. Sci. Technol., A, 1997, 15, 1710–1716 CAS
- R. Rozada, J. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. D. Tascón, Nano Res., 2013, 6, 216–233 CrossRef CAS
- S. Villar-Rodil, J. I. Paredes, A. Martinez-Alonso and J. M. D. Tascon, J. Mater. Chem., 2009, 19, 3591–3593 RSC
- A. Felten, I. Suarez-Martinez, X. Ke, G. Van Tendeloo, J. Ghijsen, J.-J. Pireaux, W. Drube, C. Bittencourt and C. P. Ewels, ChemPhysChem, 2009, 10, 1799–1804 CrossRef CAS PubMed
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS
- W.-S. Wang, D.-H. Wang, W.-G. Qu, L.-Q. Lu and A.-W. Xu, J. Phys. Chem. C, 2012, 116, 19893–19901 CAS
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS PubMed
- R. I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, John Wiley & Sons, 1996 Search PubMed
- M.-S. Park, J.-S. Yu, K. J. Kim, G. Jeong, J.-H. Kim, Y.-N. Jo, U. Hwang, S. Kang, T. Woo and Y.-J. Kim, Phys. Chem. Chem. Phys., 2012, 14, 6796–6804 RSC
- Y. Li, F. H. Yang and R. T. Yang, J. Phys. Chem. C, 2007, 111, 3405–3411 CAS
- L. Wang, J. Zhao, L. Wang, T. Yan, Y.-Y. Sun and S. B. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 21126–21131 RSC
- M. Amrita, B. Subarna, K. M. Susanta, A. G. Olivia and M. Mano, Nanotechnology, 2008, 19, 445607 CrossRef PubMed
- A. Lueking and R. T. Yang, J. Catal., 2002, 206, 165–168 CrossRef CAS
- M. Choucair and P. Mauron, Int. J. Hydrogen Energy, 2015, 40, 6158–6164 CrossRef CAS
- L. Zubizarreta, J. A. Menéndez, J. J. Pis and A. Arenillas, Int. J. Hydrogen Energy, 2009, 34, 3070–3076 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00944a |
| This journal is © The Royal Society of Chemistry 2016 |