
Formation mechanism of plasmonic silver nanohexagonal particles made by galvanic displacement reaction
Y. S. Yamamoto*ab,
Y. Fujimec,
N. Takahashi*c,
S. Nakanishia and
T. Itoh*d
aDepartment of Advanced Materials Sciences, Faculty of Engineering, Kagawa University, Takamatsu, Kagawa 761-0396, Japan. E-mail: yamayulab@gmail.com
bResearch Fellow of the Japan Society for the Promotion of Science, Chiyoda, Tokyo 102-8472, Japan
cDepartment of Physics, Faculty of Education, Kagawa University, Takamatsu, Kagawa 760-8522, Japan
dNano-Bioanalysis Research Group, Health Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Takamatsu, Kagawa 761-0395, Japan. E-mail: tamitake-itou@aist.go.jp
First published on 16th March 2016
Abstract
The galvanic displacement reaction (GDR) is a powerful method for the preparation of various plasmonic nanostructures within several minutes. However, the formation mechanism of the nanostructures, which retain plasmonic hotspots, still remains unclear. In this work, X-ray photoelectron spectroscopy (XPS) is applied to silver nanostructures made by newly-discovered GDR between Ag complex solutions and Cu alloy substrates, whose nanostructures form characteristic nanoscale hexagonal columns (NHCs) and generate strong surface-enhanced Raman scattering (SERS) signals of adsorbates. Detailed depth profiles by multi-element XPS analysis revealed that NHCs are made of Ag metallic cores covered with surface layers composed of copper sulfates and copper oxides, which prevent NHCs from fusion, resulting in highly concentrated stable hotspots. These findings explain why NHCs exhibit reproducible SERS signals and the proposed methodology gives new insights for efficient creation of plasmonic nanostructures in a few minutes.
Introduction
Numerous preparation methods for metallic nanostructures have been developed1–3 because their plasmon resonance, i.e., the coupling of collective oscillations of conducting electrons in the nanostructures with incident light generates unique plasmonic properties,4,5 which can be extensively applied to surface-enhanced spectroscopy and its expansion,6–10 acceleration of photo-catalysis,11 and photo-thermotherapy.12 The control of the plasmonic nanostructures is highly attractive1–3 because the spectral shapes and wavelengths of plasmon resonance are determined by the morphology of the nanostructures. The galvanic displacement reaction (GDR) is one of the most convenient techniques for the production of such nanostructures and has been applied to various surface-enhanced spectroscopies, e.g., surface-enhanced Raman scattering (SERS).13–23 The advantages of the GDR are its high cost-effectiveness, rapidness, and simplicity in the production processes13–23 as compared with other methods, in particular, lithography methods.1,2 Indeed, GDR-based methods are basically simple; they involve only the addition of a solution containing metal ions onto another metal surface.13However, the drawback of GDR-based methods is the difficulty in controlling both the formation and optimization of the nanostructures, resulting in a quite low reproducibility of SERS signals as compared with that of other well-controlled methods.24,25 Thus, a detailed analysis of the formation mechanism of the nanostructures is important to improve GDR-based methods for representative plasmonic applications. To clarify the mechanism underlying the formation of the nanostructures, element analysis in the depth direction is a valuable approach. X-ray photoelectron spectroscopy (XPS)26 is a powerful tool for this purpose, because the short mean free pass of photoelectrons (<several nanometers) enables the multi-element depth profile analysis with a resolution of several angstroms.27,28 In addition, XPS spectra reveal the chemical states of the multi-elements by electronic bonding energies in the nanostructures.29
In this work, we select an interesting GDR method, which was newly discovered in 2013.13 By this GDR, the nanostructures called nanoscale hexagonal columns (NHCs) can be prepared within several minutes just by the addition of thiosulfate–Ag complexes on phosphor bronze substrates and shows SERS activity.13 Usually, metal nanostructures produced by GDR methods fuse to each other, resulting in disappearance of hotspots. The hotspots, which are the crevasses generating strong SERS signal, are essential for generating high SERS signals e.g. with an enhancement factor of 108 to 1012;6,30–32 therefore, the disappearance of the hotspots results in lower SERS signals. However, interestingly, the NHCs were found to not fuse to each other and to maintain a high signal enhancement. Thus, in order to clarify this intriguing aspect, the outer surface and internal structures of the NHCs were investigated by XPS depth profile analysis of the NHCs substrates. This analysis revealed that several elements included in the NHCs exhibit different profiles, indicating that NHCs have specific internal structures. The chemical state analysis by XPS shows that the NHCs are covered with thin conductive layers and are present on the Cu depletion layer of the phosphor bronze substrates.
Experimental
NHCs on phosphor bronze substrate were synthesized by GDR, as reported elsewhere.13 Briefly, to a solution of Na2S2O3·5H2O (0.45 g) in 2 mL of H2O, AgCl (0.13 g) was added, and the mixture was stirred until dissolution was complete. The final volume was adjusted to 100 mL using H2O. In order to form the NHCs, the surface of the phosphor bronze substrate (JIS H3110, C5191P; composition of Cu + Sn + P > 99.5%, Sn 5.5–7.0%, P 0.03–0.35%, ZnO 0.20%, PbO 0.05%, and FeO 0.10%) was scratched with a diamond pen to refresh the surface. Next, the solution containing thiosulfate–Ag complexes [Ag(S2O3)n]−(2n−1) was poured on the substrate, left for 120 min, and then blown off to terminate the reaction. Upon addition of the solution, the color of the phosphor bronze substrate turned deep brown from the original metallic bronze color (inset of Fig. 1B1), thus facilitating the monitoring of the GDR process. | ||
| Fig. 1 (A1–B2) SEM images of the NHCs synthesized on the phosphor bronze substrate after (A1, A2) 3 min and (B1, B2) 120 min of reaction. Sample (B) was chosen for the XPS measurements. Inset of (B1): photograph of the phosphor bronze substrate after reaction. (C, D) Size distribution of NHCs after (C) 3 min and (D) 120 min of reaction. (E, F) Proposed schematics of the NHCs after (E) 3 min and (F) 120 min of reaction. | ||
The shapes of the NHCs were measured using a field emission scanning electron microscope (FE-SEM, JEOL, JSM-6700FZ) operating at 15 kV. XPS analyses were carried out on PHI 5000 Versaprobe (Ulvac-Phi Inc.), operating at 10−8 Pa with monochromated Al Kα radiation (photon energy 1486.6 eV).13 Prior to the XPS measurements, the surface of the NHCs was cleaned by sputtering with an Ar+ ion source of 500 eV for 1 min in order to remove oxygen sourced from the air. The conditions for recording broad XPS spectra, namely survey spectra, were as follows: 25 scans with 1 min per spectrum at a pass energy of 117.4 eV. The conditions for recording narrow XPS spectra were as follows: 100 scans with 40 s per spectrum at a pass energy of 23.5 eV. In order to obtain the XPS depth profiles, the surface of the NHCs was repeatedly sputtered by a series of 10 s bursts of Ar+ ion beam with 500 eV to expose the new subsurface, and the XPS spectra of each subsurface were measured in sequence. The resolution of the depth profiles was estimated by direct measurement of the standard samples, GaAs, under conditions identical to those used in the current experiments. The GaAs surfaces were sputtered by 1.00 nm per min with an Ar+ ion source of 500 eV; thus, the sputtering times 1 min corresponds approximately to the depth 1 nm. The X-ray beam was focused on an area with a diameter of 10 μm to collect the XPS signals only from inside the scratch occurring on the substrate. The XPS spectra were analyzed by Igor Pro software ver. 4.0.6.1 using a conventional curve fitting procedure. The X-ray diffraction (XRD) patterns were recorded using a Philips PW1710 diffractometer with the Cu KR radiation (λ = 1.54056 Å) at a scanning rate of 2 degrees per min in 2θ ranging from 20° to 120°. The whole phosphor bronze substrates partly covered with NHCs were supported on glass substrates for XRD measurements.
Results and discussion
XPS analysis is a powerful technique to reveal the detailed chemical states of elements in the nanostructures at a resolution of several angstroms along the depth direction.26–28 Indeed, XPS has been successfully applied to characterize the internal nanostructures.33 However, the resolution of XPS analysis in the lateral direction, performed by focusing the area of the X-ray beam, is of several μm2 for the light collection optical system used. Thus, in order to avoid complexity in the analysis, the phosphor bronze surface needs to be completely covered with NHCs in the micrometer order. Fig. 1A1 and A2 show the typical SEM images of the NHCs within 3 min of GDR. The hexagonal shape of NHCs is clear after 3 min of reaction,13 however, the density of the NHCs on the phosphor bronze surface is too low to record pure XPS spectra of NHCs. According to the time-course study in ref. 13, we have concluded that the formation of Ag nanostructures is taking place along with the GDR. Therefore, in order to avoid signal contamination from the phosphor bronze surface, the reaction time was increased from 3 to 120 min and a surface completely covered with NHCs was obtained. Fig. 1B1 and B2 illustrate the typical SEM images of the NHCs within 120 min of GDR. Fig. 1B2 shows that the NHCs grow perpendicularly and overlap each other. The SEM observation indicates that, on increasing the side length l, individual NHCs grow in the lateral direction rather than in the thickness direction. Fig. 1C and D show the histograms of the side length l of NHCs for a GDR time of 3 and 120 min, respectively. The average values of l were 40 nm for a GDR time of 3 min and 69 nm for a GDR time of 120 min. These results, illustrated in Fig. 1E and F, respectively, suggest that the NHCs with a GDR time of 120 min form a single NHC layer on the phosphor bronze surfaces without vacancy, indicating that the NHC samples are suitable for XPS analysis. Note that for the better morphology analysis, high-resolution transmission electron microscopy (HRTEM) was applied to visualize the shapes of NHCs, however, we didn't succeeded it because of the thickness of the phosphor bronze substrate (∼0.3 mm).To identify the elements included in NHCs, the surfaces of NHCs are measured by XPS with large spectral windows. Fig. 2 shows the XPS survey spectrum with assignments of each element peak. The peaks in the XPS survey spectrum are attributed to Ag 3d5/2, Sn 3d5/2, Cu 2p3/2, O 1s, S 2p, and C 1s,29 indicating that the outermost surface of NHCs is composed of these atoms. The proposed reactions involved in the present GDR process are:
| Ag+ + nS2O32− ⇆ [Ag(S2O3)n]−(2n−1) |
| [Ag(S2O3)n]−(2n−1) + e− ⇆ Ag* + nS2O32− |
| Ag* → Ag0 | (1) |
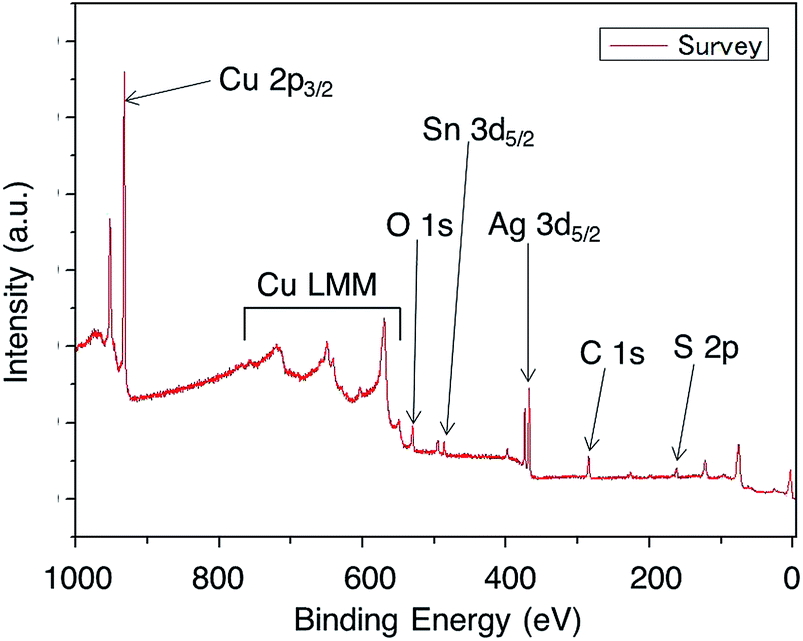 | ||
| Fig. 2 XPS survey spectra of an outermost surface of NHCs. Assignments of the spectral peaks are presented. | ||
All elements involved in eqn (1)13 are found in Fig. 2. O, Ag, and S atoms are originally contained in the thiosulfate–Ag complexes used to create NHCs. O atoms can be contaminants,34 but are also involved in the GDR process. The appearance of Cu and Sn atoms, which are present in the substrate and not included in the reaction solution, may be attributed to the dissolution of the substrate by the thiosulfate complex and its transfer to the NHC surface. C atoms are well-known contaminants of XPS,34 and will, therefore, be excluded from the discussion of the internal structures of the NHCs. The elements discussed herein are consistent with previous EDS results of NHCs.13 Thus, we conclude that Ag, Cu, Sn, O, S, and C atoms are present on the surface of the NHCs.
XPS depth profiles of Ag 3d5/2, Sn 3d5/2, Cu 2p3/2, O 1s, S 2p, and C 1s were carried out to investigate the internal structures of the NHCs. The detailed chemical states of Ag, Cu, Sn, O, S, and C atoms are analyzed from the spectral peak energies.29 Fig. 3A–F show the depth profiles of the XPS spectra of these elements. Fig. 4A–F illustrate the peak intensity plots corresponding to the XPS spectra shown in Fig. 3A–F. The unit of vertical axes of Fig. 3 and 4 was set as the “normalized photoionization cross-section”, which is proportional to the amount of atoms. That was calculated by the C 1s normalized photoionization cross-sections for Al Ka line of Ag 3d5/2 (10.66), Sn 3d5/2 (14.80), Cu 2p3/2 (16.73), O 1s (2.93), and S 2p (1.67), respectively.35
 | ||
| Fig. 3 Representative depth profiles of XPS spectra for Ag 3d5/2 (A), Sn 3d5/2 (B), Cu 2p3/2 (C), O 1s (D), S 2p (E), and C 1s (F). The sputtering time 1 min corresponds approximately to the sputtering depth 1 nm. | ||
 | ||
| Fig. 4 Area intensity plots of XPS spectra (n = 3) for Ag 3d5/2 (A), Sn 3d5/2 (B), Cu 2p3/2 (C), O 1s (D), S 2p (E), and C 1s (F), which are shown in Fig. 3. Error bars depicting ±SD exhibit spread of intensity from three independent measuring point in the same sample. The sputtering time 1 min corresponds approximately to the sputtering depth 1 nm. | ||
We herein start discussion of the amount and the chemical state of the Ag atoms in each depth. Fig. 4A shows that the XPS signal intensities of Ag 3d5/2 rapidly increase (0–10 min), saturate (10–60 min), and slowly decrease (60–100 min), indicating that the amount of Ag atoms on the surface region (0–10 min) is less than the beneath of the surface (10–60 min). Fig. 3A indicates that the XPS peak of Ag 3d5/2 at 368.3 eV does not shift and maintains a Gaussian line shape during the intensity change, suggesting that Ag atoms do not bond to other elements and exist in NHCs in the Ag0 state at every depth.29 In addition, Fig. 4A shows that the intensity profile of Ag 3d5/2 is different from that of other elements; thus, clearly, Ag atoms exist independently in the metal state without bonding to other elements. The existence of metallic Ag, which has rich conduction electrons resulting in strong plasmonic resonance,30–32 is consistent with the strong SERS signal generated from the molecules adsorbed on the NHCs.13 For further confirmation of the Ag atoms in the metal state, the XRD patterns (Fig. 5) were taken. Larger peaks at 43.1, 50.1 and 73.5 degrees are assigned to Cu and Sn, which are the main contents of phosphor bronze substrate. Smaller peaks at 38.5, 45.0, 65.2 and 78.4 degrees are all assigned to Ag fcc phase,36 thus Ag atoms inside NHCs synthesized using this method existed in fcc phase. The lattice constant of Ag calculated from this XRD pattern was 4.05 Å, which is close to the reported data (4.086 Å).37 Therefore, we conclude that Ag atoms exist in the metal state inside NHCs. The SERS activity of the NHCs13 is also supports the existence of Ag atoms in the metal state.
 | ||
| Fig. 5 Representative XRD pattern of NHCs synthesized on phosphor bronze substrate. | ||
Next, the amount and the chemical state of Sn atoms are examined. Fig. 3B shows that the XPS peak of Sn 3d5/2 at 486.5 eV does not shift and exhibits a Gaussian line shape, suggesting that Sn atoms also exist in the Sn0 state without bonding to other elements.29 Fig. 4B shows that the XPS signal intensities of Sn 3d5/2 are negligible (0–40 min), increase (40–80 min), and saturate (80–100 min). Sn atoms are originally included in the phosphor bronze substrate as a metal state; thus, the increase of Sn atoms at 50 min indicates that the Ar+ ion sputtering beam reaches the surface of the phosphor bronze substrate at a depth of >50 nm. The phosphor bronze substrate gradually appears at 50–80 nm, then at 80–100 nm the beam completely reaches the substrate. Thus, the layer at a depth of >50 nm contains the original phosphor bronze substrate, and the layer at a depth of <50 nm is estimated as NHC layer generated by the GDR, then the layer between 50–80 nm can be estimated a mixed layer of the phosphobronze substrate and the NHCs generated.
The examination of the chemical state of Cu atoms is important but rather complicated, because XPS spectra vary during the depth profile of Cu atoms. Fig. 3C demonstrates that the XPS peak of Cu 2p3/2 at 932.8 eV at 0 min shifts to 932.6 eV from 3 to 100 min. Fig. 4C shows that the XPS signal intensities of Cu 2p3/2 rapidly decrease (0–10 min), remain constant (10–60 min), and increase (60–100 min). It is natural that Cu atoms detected much in the layer at a depth of >50 nm, which is estimated as the original phosphor bronze substrate, because Cu is a main component of phosphor bronze substrate. In the phosphor bronze substrate layer, Cu atoms must exist as Cu0 metal (932.61 eV in XPS spectra of Cu 2p3/2).29 However, Cu atoms are not contained in reaction solution, thus, Cu atoms detected at a depth of <50 nm, i.e. the NHC layer, should be transferred from the phosphor bronze substrate by the GDR. This can be explained by standard chemical reactions of GDR, we discuss that later. This transference also suggests the presence of a depletion layer of Cu atoms beneath the NHCs, indeed, the depletion layer can be observed as the difference between the recovery slope of Cu 2p3/2 and that of Sn 3d5/2 (Fig. 4B and C).
Considering the chemical components Cu, O and S in the GDR (eqn (1)), the possible chemical states of the Cu atoms in NHC layer are Cu0, CuO, Cu2O, CuS, and Cu2S. Their electron binding energies of Cu 2p3/2 are reported to be 932.61, 933.57, 932.43, 932.2, and 932.5 eV, respectively.29 Thus, no candidates for the peak at 932.8 eV (0 min, outermost surface in Fig. 3C), and Cu0, Cu2O, CuS, and Cu2S are candidates for the peak at 932.6 eV (3–50 min, i.e., <50 nm except the outermost surface). In order to clarify the chemical state of Cu atoms on the outermost surface (0 min), the conventional curve fitting procedure was applied to the XPS peak of Cu 2p3/2 at 932.8 eV at 0 min. As shown in Fig. 6A, the peak at 932.8 eV at 0 min was separated into two peaks, 933.4 and 932.7 eV, which can be assigned to CuO and Cu0, Cu2O, CuS or Cu2S, respectively. Thus, on the outermost surface at 0 min, CuO also exists with other unknown Cu compounds at the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (from the peak intensity ratio in Fig. 6A). Of note is that as shown in Fig. 4A and C, on the outermost surface at 0 min, the amount of Cu atoms is more than 5 times that of Ag atoms, indicating that the main content of the outermost surface is Cu atoms, not Ag atoms. This means that the Ag0 mainly forming NHCs is not naked but covered with CuO and other Cu compounds.
:2 (from the peak intensity ratio in Fig. 6A). Of note is that as shown in Fig. 4A and C, on the outermost surface at 0 min, the amount of Cu atoms is more than 5 times that of Ag atoms, indicating that the main content of the outermost surface is Cu atoms, not Ag atoms. This means that the Ag0 mainly forming NHCs is not naked but covered with CuO and other Cu compounds.
 | ||
| Fig. 6 (A) Peak fitting result of XPS spectra of Cu 2p3/2 at 0 min. (B) and (C) Reproduced depth profiles of (B) S and (C) O atoms. (♦) Numbers of S and O atoms experimentally obtained, respectively. These are the same with Fig. 4E and D, respectively. (△) and (■) Reproduced numbers of S and O atoms by assuming CuS:Cu2S:Cu2O = 1:1:2. (×) Reproduced numbers of S atoms by assuming CuS:Cu2O = 1:1. | ||
However, at a depth of <50 nm, it is difficult to identify the chemical state of the Cu atoms only from the peak position of Cu 2p3/2. Thus, we tried to estimate the chemical states of S atoms from by XPS spectra of S 2p2/3. The XPS spectra of S 2p (Fig. 3E) always exhibit two peaks, at 161.9 and 163 eV, which can be assigned to the doublet of S 2p,38 meaning no information is obtained about the chemical states of S atoms. However, S atoms are not detected from the original phosphor bronze substrate by XPS measurements (data not shown), thus, the S atoms detected at any depth in Fig. 3E must be sourced from the reaction solution as a thiosulfate ion (S2O32−). From the proposed literature,39 S2O32− forms thiosulfate–Cu complexes with Cu ions and the disproportionation of thiosulfate–Cu complexes gives both CuS and Cu2S. Thus, in the present GDR, it is considerable that S atoms sourced as a thiosulfate ion (S2O32−) forms thiosulfate–Cu complexes with Cu ions and disproportionate into both CuS and Cu2S. In addition, by the present GDR Cu ions can be generated from Cu0 metals. The detailed chemical reactions will be summarized later.
We here also examine Cu2O as one of the chemical states of the Cu atoms with analyzing of the XPS spectra of O 1s. The XPS spectra of O 1s have a peak at 530.3 eV (Fig. 3D, 0–100 min). As illustrated in Fig. 4D, the XPS signal intensities of O 1s decrease (0–20 min) and then sharply increase (40–100 min). The electron binding energies of O 1s for CuO and Cu2O are reported to be 529.68 and 530.2 eV, respectively.29 Thus, the chemical state of the O atoms at 530.3 eV is expected to be Cu2O. O atoms are not detected from the XPS spectra obtained from the original phosphor bronze substrate after the surface sputtering of twenty nm by Ar+ ion beam (data not shown); thus, the increase in the O 1s signals from 40 to 100 min indicates that O atoms sourced from reaction solution (thiosulfate ion (S2O32−)) percolate into phosphor bronze substrate then may form Cu2O. This is consistent with the electron binding energy of Cu 2p3/2 for Cu2O, which is reported to be 932.43 eV, a value close to that of Cu2S shown in Fig. 3C (932.5 eV).29
Therefore, considering the XPS spectra peaks of Cu 2p3/2, S 2p and O 1s as well as the possible chemical reactions of all the contents, chemical state of Cu atoms is estimated as CuO, CuS, Cu2S and Cu2O for 0 min and CuS, Cu2S and Cu2O for 3–100 min. To quantify the chemical state of Cu atoms, we tried to reproduce the depth profiles of S and O atom by changing the abundance ratios of CuS, Cu2S and Cu2O. Fig. 6B and C show the best reproduction as explained below. Diamonds (♦) in Fig. 5B and C indicate the numbers of S and O atoms experimentally obtained, respectively, these are the same with Fig. 4E and D, respectively. △ and ■ in Fig. 5B and C indicate the reproduced numbers of S and O atoms by assuming CuS:Cu2S:Cu2O = 1:1:2. × indicate the reproduced numbers of S atoms by assuming CuS:Cu2O = 1:1. These reproduced depth profiles (△, ■ and ×) are well-fitted to the experimental profiles (♦) from 0–50 min, indicating in the NHC layer <50 nm, CuS, Cu2S and Cu2O are included as Cu components. The reproduced depth profiles do not fit the experimental profiles >50 nm owing to the presence of substrates.
At last, the process of the NHCs formation is summarized by considering the reported chemical reactions involving common chemical components.39–41 From eqn (1), the present GDR is categorized as the electroless plating under galvanic displacement reaction involving the silver ions and copper without any reducing agents.42,43 This electroless plating is started by adding the thiosulfate–Ag complexes [Ag(S2O3)n]−(2n−1) to the phosphor bronze substrate which contains the Cu metal. The ionization tendency of Ag is lower than that of Cu, thus the Cu metal dissolves into Cu+ and Cu2+:40,41
| Ag+ + nS2O32− ⇆ [Ag(S2O3)n]−(2n−1) |
| Ag+ + e− → Ag (E0 = 0.799 V, E0 means ionization potential) |
| Cu+ + e− → Cu (E0 = 0.520 V) |
| Cu2+ + 2e− → Cu (E0 = 0.377 V) | (2) |
Then Ag+ ions are deposited onto the phosphor bronze substrate as the Ag0 metal, forming the core of NHCs.13 While, thiosulfate ions (S2O32−) forms new complexes with Cu ions as:39
| Cu+ + nS2O32− ⇆ [Cu(S2O3)n]−(2n−1) (n = 1–3) |
| Cu2+ + nS2O32− ⇆ [Cu(S2O3)n]−(2n−2) (n = 1–2) | (3) |
| 2[Cu(S2O3)−] + 3[Cu(S2O3)0] + 3H2O ⇆ 3CuS + Cu2S + 3H2SO4 + S3O62− | (4) |
The equilibrium in eqn (3) and (4) means that the S2O32− can act as a counterpart of the Cu atoms to dissolve out Cu metal as thiosulfate–Cu complexes then the thiosulfate–Cu complexes generate both CuS and Cu2S. Both CuS and Cu2S are stable products in the present reaction condition.39 H2SO4 accelerates the reactions with the generation of the Cu2+ from the Cu metal:39
| Cu + 2H2SO4 → Cu2+ + SO42− + SO2 + 2H2O | (5) |
The generation of H2SO4 and regenerate Cu ion from Cu metal is the reason why the GDR continuously proceeds and generates larger NHCs. S atoms sourced from the reaction solution as S2O32− which percolate in the substrates >50 nm as thiosulfate–Cu complexes and may also form Cu2O although detailed chemical reaction is still unclear. However, the generation of Cu2O is reasonable because of the simulation in Fig. 6B and C. The XPS spectra of O 1s having a peak at 530.3 eV, which can be assigned to Cu2O, also supports the generation of Cu2O.
In summary, the internal structure of the NHCs and their growth is illustrated in Fig. 7A. The XPS analysis revealed that the NHCs contain mainly Ag0 metal, the surface of the NHCs is covered with a thin outermost layer which consists of CuO, CuS, Cu2S and Cu2O over the second layer which consists of CuS, Cu2S and Cu2O. Cu depletion layer is also formed under the NHCs. Thus, the growing process of the NHCs is as follows: at first, seeds of NHCs are sparsely generated from Ag complexes by receiving electrons from the Cu0 metal in the substrate; the seeds then grow and overlap each other. During the process, Cu ions bind to S2O32− ions, mainly forming CuS, Cu2S and Cu2O and covering the Ag0 cores of the NHCs. Thus the main galvanic reaction equation is:
| 2Ag[S2O3]n−(2n−1) + 5Cu0SnP + 3H2O → 2Ag0 + 3CuS + Cu2S + 3H2SO4 + S3O62− + SnP (n = 2–3) | (6) |
 | ||
| Fig. 7 (A) Proposed schematics of NHCs' growth on a phosphor bronze substrate and internal structure of NHC. (B) SEM images of Ag NPs synthesized by conventional method48 (B1 and B2) and NHCs (B3 and B4) before and after continuous electron beam irradiation, respectively. | ||
The reduced Ag+ ions are immediately incorporated in the crystal lattice of Ag0. Fig. 7A also illustrates the internal structure of the NHCs. The crucial element of the XPS analysis is the presence of about the 10 nm-thin second layer on the NHCs, covered with the <3 nm-thin outermost layer. The electric resistances of CuS (105 Ωm),44 Cu2S (10−3 Ωm)45 and Cu2O (>106 Ωm)46 are much higher than that of Ag0 (10−8 Ωm).47 Thus, the Cu2S layer serves as a dielectric layer and prevents NHCs from fusing to each other, resulting in stable SERS-active junctions in the NHC aggregates.13 To further confirm the presence of the surface layer of NHCs, the dielectric breakdown induced by continuous electron beam irradiation was examined using both metallic Ag NPs, fabricated by the Lee–Meisel method,48 and NHCs. Fig. 7B displays the SEM images of Ag NPs (Fig. 7B1 and B2) and NHCs (Fig. 7B3 and B4) before and after continuous electron beam irradiation, respectively. The dielectric breakdown appears as roughened surfaces only in the NHCs in Fig. 7B4, indicating that their surfaces are covered with a dielectric layer, mainly containing CuS, Cu2S and Cu2O.
The rationale for the formation of hexagonal shapes of Ag0 is still unclear; however, several reports on CuS and Cu2S hexagonal nanoparticles49–52 imply that Ag0 nanoparticles are stabilized by the formation of hexagonal shapes, which can be easily covered with the layer including CuS and Cu2S. Indeed, hexagonal Ag0 nanoparticles are reproducibly synthesized under proper conditions.1,2,53,54
Conclusions
XPS analysis was carried out to clarify the formation mechanism of NHCs made by the newly-discovered GDR between a Ag complex solution and a Cu alloy substrate. Multi-element XPS depth profile analysis revealed that NHCs are mainly made of metallic Ag atoms principally covered with a thin surface layers composed of copper sulfates and copper oxides, which prevents NHCs from fusing. These findings explain the strong plasmonic resonance of NHCs for sensitive SERS detection. In the proposed formation mechanism, the effects of the O atoms in the atmosphere on the GDR process were neglected. Thus, in order to obtain high-quality NHCs, the preparation of NHCs under a nitrogen atmosphere, excluding atmospheric oxygen, will be designed. The XPS analysis by synchrotron radiation facility will provide more detailed information to elucidate the unknown counterparts of the Cu atoms occurring at 932.8 eV in the spectrum of Cu 2p3/2 and will help improve the quality of the NHCs for further plasmonic applications. The rationale for the formation of hexagonal shapes of Ag0 is unclear; however, several reports on CuS and Cu2S hexagonal nanoparticles49–52 imply that Ag0 nanoparticles are stabilized by the formation of hexagonal shapes, which can be easily covered with the surface layers composed of copper sulfate and copper oxide. Indeed, hexagonal Ag0 nanoparticles are reproducibly synthesized under proper conditions1,2,53,54 as well as both CuS and Cu2S forms hexagonal shape nanoparticles.49–52Acknowledgements
The authors thank Mr Yuya Kayano (Kagawa Univ., Japan) for his assistance in data analyses, figures and references formatting. We also thank Mr Naomichi Matsumoto (Kagawa Univ., Japan) and Mr Haruo Sekiguchi (Shimadzu Techno-Research Inc., Japan) for their assistance to measure XRD patterns and those data analyses. This work was supported by the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) Number 20510111, (B) Number 26286066, WAKATE (B) Number 26810013. This work was also supported by the Research Fellowship (RPD) of the Japan Society for the Promotion of Science (JSPS) Number 26-40216 and partly supported by Tanaka Precious Metals (Tanaka Kikinzoku Group) through its “Precious Metals Research Grants”.References
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669 CrossRef CAS PubMed.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736 CrossRef CAS PubMed.
- H. Liu, Z. Yang, L. Meng, Y. Sun, J. Wang, L. Yang, J. Liu and Z. Q. Tian, J. Am. Chem. Soc., 2014, 136, 5332 CrossRef CAS PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783 CrossRef CAS.
- D. Y. Wu, J. F. Li, B. Ren and Z. Q. Tian, Chem. Soc. Rev., 2008, 37, 1025 RSC.
- Y. S. Yamamoto, M. Ishikawa, Y. Ozaki and T. Itoh, Front Phys China, 2014, 9, 31 Search PubMed.
- T. Itoh, Y. S. Yamamoto, H. Tamaru, V. Biju, N. Murase and Y. Ozaki, Phys. Rev. B, 2013, 87, 235408 CrossRef.
- T. Itoh, Y. S. Yamamoto, H. Tamaru, V. Biju, S. Wakida and Y. Ozaki, Phys. Rev. B, 2014, 89, 195436 CrossRef.
- M. Takase, H. Ajiki, Y. Mizumoto, K. Komeda, M. Nara, H. Nabika, S. Yasuda, H. Ishihara and K. Murakoshi, Nat. Photonics, 2013, 7, 550 CrossRef CAS.
- Y. S. Yamamoto, Y. Ozaki and T. Itoh, J. Photochem. Photobiol., C, 2014, 21, 81 CrossRef CAS.
- K. Ueno and H. Misawa, J. Photochem. Photobiol., C, 2013, 15, 31 CrossRef CAS.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115 CrossRef CAS PubMed.
- Y. S. Yamamoto, K. Hasegawa, Y. Hasegawa, N. Takahashi, Y. Kitahama, S. Fukuoka, N. Murase, Y. Baba, Y. Ozaki and T. Itoh, Phys. Chem. Chem. Phys., 2013, 15, 14611 RSC.
- S. Mabbott, I. A. Larmour, V. Vishnyakov, Y. Xu, D. Graham and R. Goodacre, Analyst, 2012, 137, 2791 RSC.
- P. R. Brejna, U. Sahayrn, M. G. Norton and P. R. Griffiths, J. Phys. Chem. C, 2011, 115, 1444 CAS.
- A. Gutes, C. Carraro and R. Maboudian, J. Am. Chem. Soc., 2010, 132, 1476 CrossRef CAS PubMed.
- Y. Wang, M. Becker, L. Wang, J. Q. Liu, R. Scholz, J. Peng, U. Gosele, S. Christiansen, D. H. Kim and M. Steinhart, Nano Lett., 2009, 9, 2384 CrossRef CAS PubMed.
- Y. G. Sun, Adv. Funct. Mater., 2010, 20, 3646 CrossRef CAS.
- L. Su, W. Z. Jia, D. P. Manuzzi, L. C. Zhang, X. P. Li, Z. Y. Gu and Y. Lei, RSC Adv., 2012, 2, 1439 RSC.
- M. Pradhan, J. Chowdhury, S. Sarkar, A. K. Sinha and T. Pal, J. Phys. Chem. C, 2012, 116, 24301 CAS.
- S. P. Xie, X. C. Zhang, D. Xiao, M. C. Paau, J. Huang and M. M. F. Choi, J. Phys. Chem. C, 2011, 115, 9943 CAS.
- M. Bechelany, P. Brodard, J. Elias, A. Brioude, J. Michler and L. Philippe, Langmuir, 2010, 26, 14364 CrossRef CAS PubMed.
- Y. Lai, W. Pan, D. Zhang and J. Zhan, Nanoscale, 2011, 3, 2134 RSC.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- C. R. Brundle and A. D. Baker, Electron Spectroscopy: Theory, Techniques and Application, Academic Press, London, 1978, vol. 2 Search PubMed.
- C. S. Fadley, J. Electron Spectrosc. Relat. Phenom., 2010, 178–179, 2–32 CrossRef CAS.
- F. Zhang, P. Wang, J. Koberstein, S. Khalid and S. W. Chan, Surf. Sci., 2004, 563, 74 CrossRef CAS.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST Standard Reference Database 20, Version 4.1, http://srdata.nist.gov/xps/Default.aspx, accessed, June 30, 2015 Search PubMed.
- M. Inoue and K. Ohtaka, J. Phys. Soc. Jpn., 1983, 52, 3853 CrossRef CAS.
- K. Imura, H. Okamoto, M. Hossain and M. Kitajima, Nano Lett., 2006, 6, 2173 CrossRef CAS PubMed.
- K. Yoshida, T. Itoh, H. Tamaru, V. Biju, M. Ishikawa and Y. Ozaki, Phys. Rev. B, 2010, 81, 115406 CrossRef.
- D. D. Sarma, P. K. Santra, S. Mukherjee and A. Nag, Chem. Mater., 2013, 25, 1222 CrossRef CAS.
- C. E. Taylor, S. D. Garvey and J. E. Pemberton, Anal. Chem., 1996, 68, 2401 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1975, 8, 129 CrossRef.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736 CrossRef CAS.
- P. Villars, Pearson's Handbook Desk Edition, ASM International, Materials Park, OH, 1997 Search PubMed.
- J. Peterson Phillip, Corrosion of Electronic and Magnetic Materials, Astm Intl, West Conshohocken, 1992, vol. 1148 Search PubMed.
- G. Senanayake, Miner. Eng., 2005, 18, 409 CrossRef CAS.
- J. C. Ghosh, J. Indian Chem. Soc., 1974, 51, 361 CAS.
- CRC Handbook of Chemistry and Physics, Internet Version 2005, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- M. Schlesinger, in Modern Electroplating, ed. M. Schlesinger and M. Paunovic, John Wiley & Sons, Inc, Hoboken, NJ, USA, 5th edn, 2010, vol. 5, p. 131 Search PubMed.
- T. Osaka, Y. Okinaka, J. Sasano and M. Kato, STAM, 2006, 7, 425 CAS.
- S. Erokhina, V. Erokhin, C. Nicolini, F. Sbrana, D. Ricci and E. Zitti, Langmuir, 2003, 19, 766 CrossRef CAS.
- F. El Akkad, B. Mansour and T. Hendeya, Mater. Res. Bull., 1981, 16, 535 CrossRef CAS.
- F. L. Weichman and B. C. McInnis, Can. J. Phys., 1965, 43, 507 CrossRef CAS.
- R. A. Matula, J. Phys. Chem. Ref. Data, 1979, 8, 1147 CrossRef CAS.
- P. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391 CrossRef CAS.
- J. Shan, P. Pulkkinen, U. Vainio, J. Maijala, J. Merta, H. Jiang, R. Serimaa, E. Kauppinend and H. Tenhu, J. Mater. Chem., 2008, 18, 3200 RSC.
- W. Song, J. Wang, Z. Mao, W. Xu and B. Zhao, Spectrochim. Acta, Part A, 2011, 79, 1247 CrossRef CAS PubMed.
- P. Zhang and L. Gao, J. Mater. Chem., 2003, 13, 2007 RSC.
- X. Li, H. Shen, J. Niu, S. Li, Y. Zhang, H. Wang and L. S. Li, J. Am. Chem. Soc., 2010, 132, 12778 CrossRef CAS PubMed.
- G. P. Lee, A. I. Minett, P. C. Innisa and G. G. Wallace, J. Mater. Chem., 2009, 19, 8294 RSC.
- N. Cathcart and V. Kitaev, ACS Nano, 2011, 5, 7411 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |