
Retracted Article: Monodispersed palladium–cobalt alloy nanoparticles assembled on poly(N-vinyl-pyrrolidone) (PVP) as a highly effective catalyst for dimethylamine borane (DMAB) dehydrocoupling†
Abstract
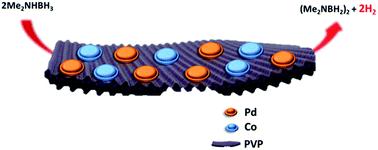
Herein we report the fabrication of monodispersed poly(N-vinyl-2-pyrrolidone) supported palladium–cobalt nanomaterials (3.45 ± 0.36 nm) and their outstanding efficiency as catalysts in dimethylamine-borane dehydrogenation. By the use of an ultrasonic double reduction method, palladium and cobalt cations were co-reduced in PVP solution and then the prepared nanocatalysts were characterized by UV-Vis, XRD, XPS and HR-TEM-EDX analyses. The nanocatalysts could be easily reused, and at extremely low concentrations and temperature they showed record catalytic activity, giving the best catalytic performance yet with a very high turnover frequency (330.94 h−1) and a low Ea value of 50.78 ± 2 kJ mol−1 for DMAB dehydrocoupling.
 Please wait while we load your content...
Please wait while we load your content...

