
Methanation of carbon monoxide on ordered mesoporous NiO–TiO2–Al2O3 composite oxides
Abstract
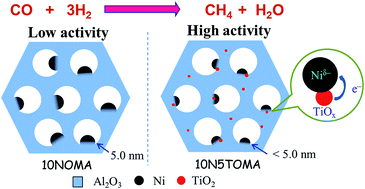
Simultaneous inhibition of the sintering of Ni particles and coke formation, while maintaining high catalytic activity, is greatly challenging for supported Ni catalysts in high-temperature applications. To address this problem, a series of ternary ordered mesoporous NiO–TiO2–Al2O3 composite oxides were synthesized via the evaporation-induced self-assembly (EISA) method and applied in the CO methanation reaction. The addition of TiO2 could significantly promote the catalytic activity, and the optimal ordered mesoporous NiO–TiO2–Al2O3 catalyst with a composition of 10 wt% NiO and 5 wt% TiO2 (10N5TOMA) achieved the maximum CH4 yield of 93% at 380 °C, 0.1 MPa and a weight hourly space velocity of 60 000 mL g−1 h−1. Our characterization results showed that the promotive effect of TiO2 appeared to be two-fold: on the one hand, the Ti species could decrease the Ni particle size and improve the reducibility of Ni particles, leading to increased H2 uptake and the dispersion of the metallic Ni; on the other hand, the electron cloud density of Ni was increased by electron transfer from Ti4+/Ti3+ redox, which might facilitate CO dissociation from the catalyst surface. In addition, the ordered mesoporous 10N5TOMA catalyst showed superior anti-sintering and anti-coking properties in a 550 °C-139 h-lifetime test, mainly because the Ni particles were anchored in the alumina matrix with a strong interaction between the Ni species and the ordered mesoporous alumina (OMA) framework.
 Please wait while we load your content...
Please wait while we load your content...

