
In situ generation and trapping of thioimidates: an intermolecular tandem reaction to 4-acylimino-4H-3,1-benzothiazines†
Christian Steinebacha,
Anna-Christina Schulz-Finckea,
Gregor Schnakenburgb and
Michael Gütschow*a
aPharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn, An der Immenburg 4, D-53121 Bonn, Germany. E-mail: guetschow@uni-bonn.de; Fax: +49 228 732567; Tel: +49 228 732317
bInstitute of Inorganic Chemistry, University of Bonn, Gerhard-Domagk-Strasse 1, D-53121 Bonn, Germany
First published on 29th January 2016
Abstract
The proton-catalyzed transformation of 2-thioureidobenzonitriles to 4-acylimino-4H-3,1-benzothiazines was accomplished by treatment with carboxylic anhydrides, acid chlorides or alkyl chloroformates. The reaction involves a cyclization to 4-imino-3,1-benzothiazinium salts, whose thioimidate structure is trapped by a subsequent reaction with the acyl donor. 2-Ureidobenzonitriles do not undergo such an intermolecular tandem reaction. The different reaction behavior of both types of substrates was verified by NMR monitoring and employing 18O-enriched water.
Introduction
The reaction of carbonitriles with sulfur nucleophiles has attracted much attention, in particular for a variety of heterocyclizations. Moreover, this transformation was successfully utilized for the design of peptide nitriles and azapeptide nitriles as inhibitors of cysteine proteases. Such nitrile-based peptides can be obtained by replacing the scissile peptide bond of a specific protease substrate by the cyano warhead and additional peptidomimetic modifications.1,2 Odanacatib, an inhibitor of the therapeutically relevant cysteine protease cathepsin K, is likely to be registered for clinical use against osteoporosis.3 The covalent reaction of peptide nitriles with cysteine proteases involves the attack of the active-site thiolate at the nitrile carbon and the reversible thioimidate formation.1,2 Thioimidates are amenable to N-acylation, a transformation that has been performed for acyclic and cyclic thioimidate substrates.4,5This study was aimed at investigating a trapping reaction of cyclic thioimidates or imidates through an intermolecular tandem reaction with acyl donors. As substrates for the envisaged ring closure reaction, 2-thioureidobenzonitriles 1 and 2-ureidobenzonitriles 2, respectively, were chosen (Fig. 1). These carbonitriles possess the sulfur or oxygen atom as part of the tautomerizable thiourea or urea moiety, which is attached at the position adjacent to the cyano group. Due to the bis-alkylated terminal nitrogen of 1 and 2, a heterocyclization is only possible via the sulfur or oxygen and would produce a fused six membered ring leading to 4H-3,1-benzothiazines and 4H-3,1-benzoxazines, respectively.6–8 Moreover, the conversion of 2-thioureidobenzonitriles 1 to 2-amino-4H-3,1-benzothiazin-4-ones 7 (R1, R2 ≠ H),9,10 which occurred upon heating in concd HCl was expected to proceed via cationic thioimidates of type 3 and/or 5. Such intermediates could undergo a trapping reaction in the presence of appropriate acyl donors.
 | ||
| Fig. 1 2-(Thio)ureidobenzonitriles and derived heterocyclic structures. | ||
Representatives of the heterocyclic class of 2-amino-4H-3,1-benzothiazin-4-ones, that is, compounds 7 (Fig. 1), possess dual activities as adenosine receptor antagonists and monoamine oxidase B inhibitors,12 and act as ligands of the oxoeicosanoid receptor (OXE-R).13–15 The formal replacement of the ring sulfur in 7 by oxygen leads to 2-amino-4H-3,1-benzoxazin-4-ones 8 (Fig. 1). These heterocycles are intrinsically less stable, as it has been shown by nonenzymatic hydrolysis.9,16 Thus, 4H-3,1-benzoxazin-4-ones are susceptible to a nucleophilic attack by the active-site serine residue of serine proteases. As peptide substrates, the benzoxazinone inhibitors interact with serine proteases in the course of an acyl transfer reaction, leading to the formation of an acyl enzyme, which undergoes hydrolytic deacylation. Inspired by this mechanism, 2-amino- and 2-alkoxy-4H-3,1-benzoxazin-4-ones have been developed as highly potent inhibitors of therapeutically relevant serine proteases, such as human leukocyte elastase, cathepsin G, chymase, C1r serine protease of the complement system and human cytomegalovirus protease.17–20
Herein we report on an intermolecular tandem reaction to 4-acylimino-4H-3,1-benzothiazines 11–16 (Fig. 2) upon treatment of 2-thioureidobenzonitriles 1 with different acyl donors under acidic conditions. The reaction includes the ring closure of 1 to iminothiazinum salts 3, the imino acylation to form the salts 9 and their deprotonation to 11–16. In contrast, a corresponding transformation of 2-ureidobenzonitriles 2 via intermediate salts 4 and 10 did not occur and 4-acylimino-4H-3,1-benzoxazines were not obtained.
 | ||
| Fig. 2 Synthesis of 4-acylimino-4H-3,1-benzothiazines 11–16. (a) CSCl2, CH2Cl2, H2O, rt, 3 h; (b) HNR1R2, CH2Cl2, rt, 1 h; (c) (i) (R3CO)2O, concd H2SO4 or (R3CO)2O, MeCN, concd H2SO4 or R3COCl, MeCN, concd H2SO4; (ii) H2O, NaHCO3, pH = 7; (iii) EtOAc; (d) ClCO2Ph, toluene, reflux, 2 h; (e) HNR1R2, THF, reflux, 2 h; (f) (i) Ac2O, concd H2SO4, rt, 2 h; (ii) H2O, NaHCO3, pH = 7; (iii) EtOAc. | ||
Results and discussion
As initial step of this study, we prepared the substrates for the envisaged generation of cyclic thioimidates and imidates. The 2-thioureidobenzonitriles 1a and 1b,21 as well as 1c (Fig. 2) were obtained by the reaction of 2-isothiocyanatobenzonitrile with diethylamine, morpholine and N-benzylmethylamine, respectively. As expected,22 the formation of analogous urea substrates 2 through an imidazole-carbonylation of the deactivated anthranilonitrile followed by treatment with secondary amines raised difficulties. However, a synthetic entry was achieved by converting anthranilonitrile with phenyl chloroformate to the corresponding carbamate,23 which was subsequently reacted with the aforementioned amines to the new 2-ureidobenzonitriles 2a–c.24In order to estimate whether thioureas 1 undergo a proton-catalyzed ring closure, we dissolved 1a in CD3CO2D and subjected the solutions to NMR analysis (see also ESI, Fig. S1 and S2†). A mixture of two compounds was observed after two hours of incubation. The minor component (40%, based on 1H NMR peak integration) was the educt 1a showing 1H and 13C NMR resonances highly similar with those obtained in a different solvent, i.e. DMSO-d6. Noteworthy, the NMR signals of the major component (60%), indicated the formation of a cyclized compound. Its 3,1-benzothiazine structure was assumed when considering the absence of a 13C NMR nitrile resonance and comparing the observed aromatic 1H and 13C signals with those of 2-amino-4H-3,1-benzothiazin-4-ones 7 (structure in Fig. 1).12,13 The treatment of 1a with AcOH (Fig. 3) in a preparative scale formed a solution whose yellow color was attributed to the formation of the cyclized 4-iminothiazinium salt, either protonated at the 2-amino substituent (3a) or the 4-imino nitrogen (5a). However, after dilution with water and extraction of the neutralized solution, the starting material, compound 1a, was quantitatively re-obtained. Hence, we concluded that 1a undergoes a reversible, proton-catalyzed ring closure to 3a. The ring opening reaction of 3a resembles the known behavior of thioimidates prone to generate nitriles in the course of the elimination of the corresponding thiol.4,5 In contrast, 2-acylamino-4-imino-4H-3,1-benzothiazines have been isolated as cyclized products.11
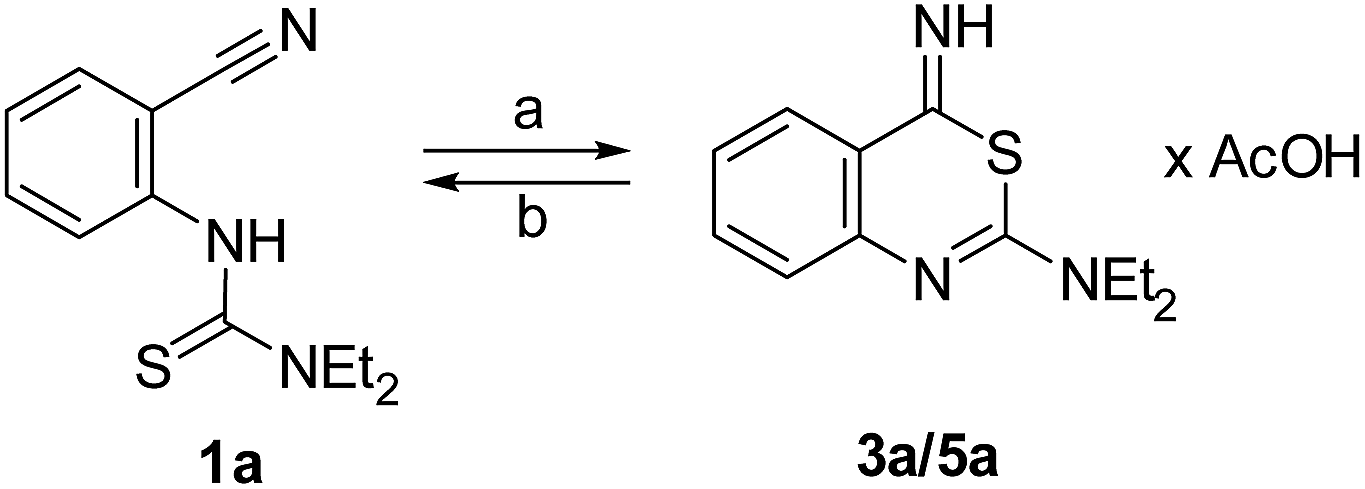 | ||
| Fig. 3 Reversible formation of a cyclic thioimidate from 1a. (a) AcOH, rt, 2 h; (b) (i) H2O, NaHCO3, pH = 7; (ii) CH2Cl2. | ||
The treatment of the selected urea derivative 2b with CD3CO2D, under the same conditions as used before for the thiourea 1a, did not provide an indication for an oxazine ring closure. NMR signals for iminooxazinium salts of type 4 or 6 (structures in Fig. 1) were not observed. We detected only 1H and 13C NMR resonances of the starting 2-ureidobenzonitrile (see also ESI, Fig. S3 and S4†). Thus, the urea and thiourea substrates behaved differently. The higher polarizability of the sulfur atom and a better resonance stabilization of a resulting thiazine ring was assumed to account for a preferred cyclization of the 2-thioureidobenzonitrile. The urea 2b was then also treated with AcOH in a preparative scale. As expected, no conversion was observed after work-up.
These results prompted us to choose the thiourea educts and study a possible trapping reaction of the 2-amino-4-imino-4H-3,1-benzothiazine intermediates by promoting an intermolecular cascade reaction, which was envisaged to occur when acyl donors were added to the reaction mixture. For this purpose, the thiourea 1a was heated in a mixture of propionic anhydride and concd H2SO4 (Fig. 2). At once, the solution took on a red color which remained until the end of the 4 hours reaction time. Attempts to extract an organic product with ethyl acetate after pouring the reaction solution onto ice-water failed. However, we succeeded when first neutralizing the ice-cold aqueous mixture followed by extraction with ethyl acetate. Preparative column chromatography provided the yellow propionylimino derivative 12a in 81% yield. Its structure was confirmed by an X-ray crystal analysis (Fig. 4).25
 | ||
| Fig. 4 Molecular plot of 2-diethylamino-4-propionylimino-4H-3,1-benzothiazine (12a) showing the displacement ellipsoids at the 30% probability level for the non-H atoms. H atoms are depicted as white spheres of arbitrary radii.25 | ||
The mechanism of this conversion was envisaged to include a proton-catalyzed activation of the cyano group, nucleophilic attack of the sulfur and ring closure leading to the reversible formation of a benzothiazinium salt 3 (Fig. 1). This intermediate is susceptible to a trapping reaction in the presence of the acyl donor propionic anhydride, leading to the formation of the red, water soluble acyliminium salt 9. If 9 gets deprotonated in the course of work-up, the yellow acylimino product 12 can be isolated (see also ESI, Fig. S5†).26
By applying this protocol for the intermolecular tandem reaction, besides 12a, eight further 2-amino-4H-3,1-benzothiazines with 4-acetylimino (11a–c), 4-propionylimino (12b, c), and 4-isobutyrylimino (13a–c) residues were successfully produced (Fig. 2). It turned out that a prolonged reaction time (2 h for 11, 4 h for 12 and 6 h for 13) and stronger conditions (50 °C for 11 and 12, 60 °C for 13) were needed to account for the decreased reactivity and increased viscosity of the corresponding carboxylic anhydrides. Anhydrides with higher melting points were not suitable as reaction medium. To overcome this obvious disadvantage of the method, we modified the composition of the reaction mixture and treated 1a with propionic anhydride and acetonitrile as solvent in the presence of concd H2SO4. Actually, this reaction again yielded 12a and the conditions were applied to introduce further acyl residues. With isovaleric anhydride in acetonitrile, the acylimino products 14a–c were synthesized. Chloroacetic anhydride was employed for the preparation of the derivatives 15a–c.
In an exemplary reaction, when using propionyl chloride instead of propionic anhydride, as well as acetonitrile and concd H2SO4, 1a was again converted to 12a. This experiment demonstrated the applicability of acid chlorides to serve as acyl donors in the course of the formation of 4-acylimino-4H-3,1-benzothiazines. Next, we examined whether cyclic thioimidates can also be trapped with chloroformates. The reactions of thioureas 1a–c with benzyl chloroformate furnished the final compounds 16a–c, with a carbamate-type 4-imino substituent. In these preparations, excess benzyl chloroformate and benzyl alcohol were removed by a first column chromatographic step prior to the evaporation of the solvent.27
The structure type 16 was investigated with respect to a possible hydrogenolytic deprotection of the imino moiety. If so, it might be verified whether the non-protonated cyclized iminothiazine would be stable in neutral media or would spontaneously undergo ring opening to a thioureidobenzonitrile 1. Initially, we tested different hydrogenolytic conditions to assure that the benzothiazine skeleton would not be affected and which could then be applied to the cleavage of the benzyloxycarbonyl group. The representative 12a remained unchanged when subjected to a palladium-catalyzed hydrogenation (Pd/C 10 wt%, H2, 3 atm) over 4 h in THF at room temperature. However, the Z-protected derivative 16a was also stable under these conditions. We attribute this resistance of the carbamate moiety towards catalytic hydrogenolysis to the poisoning effect of the sulfur atom.28
In order to confirm unequivocally the structure of 4-acylimino-4H-3,1-benzothiazines deduced from their 1H and 13C NMR spectra and to assign the configuration of the acyl residue relative to the benzothiazine-skeleton, a single crystal X-ray analysis was carried out on the propionylimino derivative 12a (Fig. 4).25 The central benzothiazine ring system is planar with a maximum deviation from planarity of 0.092 Å. The favorable Z-configuration of the C(4)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(3) double bond can be explained by assumption of polar interactions between the S and O atoms, whose nonbonded intramolecular distance of 2.61 Å was less than the sum of the van der Waals radii of 3.32 Å. The atoms S, C(4), N(3), C(13) and O also form a plane with a maximum deviation from planarity of 0.057 Å which is coplanar with the fused ring system.
N(3) double bond can be explained by assumption of polar interactions between the S and O atoms, whose nonbonded intramolecular distance of 2.61 Å was less than the sum of the van der Waals radii of 3.32 Å. The atoms S, C(4), N(3), C(13) and O also form a plane with a maximum deviation from planarity of 0.057 Å which is coplanar with the fused ring system.
The 2-ureidobenzonitriles 2a–c were also studied as substrates for the intermolecular tandem reaction (Fig. 2). The treatment with acetic anhydride in the presence of concd H2SO4 followed by dilution with water and extraction of the neutralized aqueous solution produced mixtures of the corresponding educts 2a–c and 4H-3,1-benzoxazin-4-ones 8a–c. The latter products were isolated and their structure was unambiguously confirmed by NMR and LC/MS data and by comparison with independently prepared 8a–c.18,29 The unexpected formation of 4H-3,1-benzoxazin-4-ones 8a–c from 2a–c was assumed to result from hydrolysis of either a 4-imino-3,1-benzoxazine salt or a 4-acylimino-3,1-benzoxazine. To shed light on the mechanism of the formation of 8, we performed a back-to-back experiment and subjected the 2-ureidobenzonitrile 2c to the conditions of the intermolecular tandem reaction. The work-up was either done with a solution of NaHCO3 in H2O or in 18O-enriched water. After extraction with ethyl acetate and purification by column chromatography, both 4H-3,1-benzoxazin-4-one products were analyzed by HRMS. However, work-up with H218O did not result in an incorporation of 18O into the molecule, which would have been the case when a 4-imino-3,1-benzoxazine salt or a 4-acylimino-3,1-benzoxazine were hydrolyzed to produce 8. These results clearly indicate that 2-ureidobenzonitriles 2, under conditions applied herein, do not generate 4-acylimino-3,1-benzoxazines. Most likely, the formation of the cyclized products 8 from 2 occurred before the aqueous work-up and needs further investigations.
While 4H-3,1-benzothiazin-4-ones 7 were only weak protease inhibitors,9 an oxygen-acylimino replacement might lead to an altered reactivity and a changed biological activity. Thus, we aimed at studying the interaction of 4-acylimino-4H-3,1-benzothiazines 11–16 with serine and cysteine proteases. The mechanism by which these enzymes cleave their substrates includes the nucleophilic attack of the active site serine oxygen or cysteine sulfur at the carbon of the scissile peptide bond and the formation of an acyl-enzyme. A covalent interaction is also typical for inhibitors of serine and cysteine proteases. Compounds 11–16 were not active against the serine protease chymotrypsin and the cysteine proteases cathepsin B and cathepsin L, where they showed IC50 values of more than 10 μM. The serine protease human leukocyte elastase (HLE) was inhibited by several 4-acylimino-4H-3,1-benzothiazines, bearing an aromatic moiety within the 2-substitutent or within the 4-substituent. Combination of both features provided the most potent HLE inhibitor, compound 16c, with an N-benzyl(methyl)amino group at position 2 and a benzyloxycarbonylimino group at position 4 (IC50 = 3.17 μM, see ESI, Table S3†). Derivatives with a 4-chloroacetylimino group did not inhibit HLE, indicating electronic parameters not to be crucial for HLE inhibition. Since, moreover, the inhibition was not time-dependent, we did not assume a covalent mode of interaction.
Conclusions
An efficient intermolecular tandem reaction for the preparation of trapped thioimidates was developed, which might be applicable also to other types of substrates, for example with a thioamide or thiocarbamate moiety at the position adjacent to the cyano group. Moreover, other acyl donors, such as electrophilic sulfur or phosphorus compounds might be employed. The resulting 4H-3,1-benzothiazines, together with the products of this study, appear to be worthwhile for further biological evaluations at different targets.Experimental
General experimental methods
Reagents were obtained from Acros, Aldrich, Alfa Aesar, Bachem, Carbolution and Fluorochem. 2-Isothiocyanatobenzonitrile was available from 2-aminobenzonitrile and thiophosgene.30 Thin-layer chromatography was carried out on aluminum sheets, coated with silica gel 60 F254 (Merck). The corresponding retention factors are noted below. Compounds were visualized under UV light (254 nm). Preparative column chromatography was performed using Merck silica gel 60 (63–200 mesh). Petroleum ether used was a mixture of alkanes boiling between 40–60 °C, according to the supplier's declaration. Melting points were determined on a Büchi 510 oil bath apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 500 MHz NMR spectrometer and on a Bruker Avance III 600 MHz NMR spectrometer, respectively. NMR spectra were recorded in DMSO-d6 at 303 K or CD3CO2D at 298 K. Chemical shifts are given in parts per million (ppm) referring to the signal center using the solvent peaks for reference: DMSO-d6 2.49/39.7 ppm and CD3CO2D 2.04, 11.65/20.0, 178.99. Coupling constants J are given in Hertz, and spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), hept (heptet) and non (nonet). HRMS was recorded on a micrOTOF-Q mass spectrometer (Bruker) with ESI-source coupled with an HPLC Dionex Ultimate 3000 (Thermo Scientific) using an EC50/2 Nucleodur C18 Gravity 3 μm column (Macherey-Nagel). A volume of 1 μL of a sample solution 1.0 mg mL−1 was injected. Mobile phase was water containing 2 mM ammonium acetate/acetonitrile. Elution was performed from 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 up to 0:100 in 9 min, 0:100 for 5 min. Elemental analyses were performed with a Vario EL apparatus for C, H, and N. The purity of the compounds was determined by HPLC-UV obtained on an LC-MS instrument (Applied Biosystems API 2000 LC/MS/MS, HPLC Agilent 1100) using the procedure as follows: dissolving of the compounds at a concentration of 1.0 mg mL−1 in MeCN and if necessary sonicated to complete dissolving. Then, 10 μL of the substance solution was injected into a Phenomenex Luna C18 HPLC column (50 × 2.00 mm, particle size 3 μm) and elution performed with a gradient of water/MeOH either containing 2 mM ammonium acetate from 90:10 up to 0:100 for 30 min at a flow rate of 300 μL min−1, starting the gradient after 1 min. UV absorption was detected from 220 to 400 nm using a diode array detector, unless noted otherwise.
:1); 1H NMR (500 MHz, DMSO-d6) δ 7.15–7.61 (m, 7H, 3-H, 5-H, 2′-H, 3′-H, 4′-H), 7.68–7.75 (m, 1H, 4-H), 7.84 (dd, J = 7.8, 1.5 Hz, 1H, 6-H), 10.32 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 108.04 (C-1), 116.87 (CN), 121.83, 125.72, 125.76, 126.19, 129.60 (C-3, C-5, C-2′, C-3′, C-4′), 133.52, 134.10 (C-4, C-6), 140.09, 150.67, 152.51 (C-1′, C-2, CO).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.12 (t, J = 7.0 Hz, 6H, CH3), 3.34 (q, J = 7.1 Hz, 4H, CH2), 7.21–7.46 (m, 2H, 3-H, 5-H), 7.58–7.61 (m, 1H, 4-H), 7.70 (dd, J = 7.8, 1.6 Hz, 1H, 6-H), 8.44 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 13.89 (CH3), 40.89 (CH2), 108.22 (C-1), 117.46 (CN), 124.30, 125.59 (C-3, C-5), 132.71, 133.30 (C-4, C-6), 142.98 (C-2), 154.42 (CO); LC-MS (ESI) tR = 6.54 min, 99% purity, m/z [M + H]+ calcd for C12H15N3O, 218.28; found, 218.3. HRMS-ESI m/z [M + H]+ calcd for C12H15N3O, 218.1288; found, 218.1295.:2); 1H NMR (600 MHz, DMSO-d6) δ 3.42–3.47 (m, 4H, NCH2), 3.59–3.64 (m, 4H, OCH2), 7.24–7.42 (m, 2H, 3-H, 5-H), 7.60–7.63 (m, 1H, 4-H), 7.73 (dd, J = 7.7, 1.6, Hz, 1H, 6-H), 8.86 (s, 1H, NH); 13C NMR (151 MHz, DMSO-d6) δ 44.57 (NCH2), 66.10 (OCH2), 108.34 (C-1), 117.40 (CN), 124.66, 125.46 (C-3, C-5), 132.97, 133.53 (C-4, C-6), 142.63 (C-2), 155.23 (CO); LC-MS (ESI) tR = 3.89 min, 99% purity, m/z [M + H]+ calcd for C12H13N3O2, 232.10; found, 232.2. HRMS-ESI m/z [M + H]+ calcd for C12H13N3O2, 232.1081; found, 232.1074.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.92 (s, 3H, CH3), 4.56 (s, 2H, CH2), 7.24–7.48 (m, 7H, 3-H, 5-H, 2′-H, 3′-H, 4′-H), 7.58–7.66 (m, 1H, 4-H), 7.73 (dd, J = 7.7, 1.7 Hz, 1H, 6-H), 8.73 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 34.26 (CH3), 51.41 (CH2), 108.54 (C-1), 117.49 (CN), 124.59, 125.50 (C-3, C-5), 127.18, 127.47, 128.57 (C-2′, C-3′, C-4′), 132.90, 133.45 (C-4, C-6), 138.10 (C-1′), 142.78 (C-2), 155.74 (CO); LC-MS (ESI) tR = 7.84 min, 99% purity, m/z [M + H]+ calcd for C16H15N3O, 266.32; found, 266.3. HRMS-ESI m/z [M + H]+ calcd for C16H15N3O, 266.1288; found, 266.1282.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.18 (t, J = 7.0 Hz, 6H, CH2CH3), 2.32 (s, 3H, COCH3), 3.58 (q, J = 7.1 Hz, 4H, CH2), 7.13–7.17 (m, 1H, 6-H), 7.25 (dd, J = 8.2, 1.1 Hz, 1H, 8-H), 7.58–7.60 (m, 1H, 7-H), 8.11 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.04 (CH2CH3), 26.31 (COCH3), 43.13 (CH2), 113.92 (C-4a), 123.34, 124.84 (C-6, C-8), 127.81 (C-5), 135.10 (C-7), 149.34 (C-8a), 152.02 (C-2), 158.72 (C-4), 183.39 (CO); LC-MS (ESI) tR = 13.77 min, 97% purity, m/z [M + H]+ calcd for C14H17N3OS, 276.11; found, 276.1. HRMS-ESI m/z [M + H]+ calcd for C14H17N3OS, 276.1165; found, 276.1157.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3), 3.61–3.73 (m, 8H, CH2), 7.18–7.25 (m, 1H, 6-H), 7.28 (dd, J = 8.4, 1.2 Hz, 1H, 8-H), 7.60–7.68 (m, 1H, 7-H), 8.13 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 26.26 (CH3), 45.69 (NCH2), 65.72 (OCH2), 114.41 (C-4a), 124.18, 124.93 (C-6, C-8), 127.89 (C-5), 135.21 (C-7), 148.46 (C-8a), 153.58 (C-2), 157.99 (C-4), 183.38 (CO); LC-MS (ESI) tR = 11.45 min, 98% purity, m/z [M + H]+ calcd for C14H15N3O2S, 290.09; found, 290.1. Anal. calcd for C14H15N3O2S: C, 58.11%; H, 5.23%; N, 14.52%. Found: C, 58.01%; H, 5.32%; N, 14.37%.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.31 (s, 3H, COCH3); 3.14 (s, 3H, NCH3), 4.88 (s, 2H, CH2), 7.15–7.23 (m, 1H, 6-H), 7.23–7.40 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.59–7.66 (m, 1H, 7-H), 8.15 (dd, J = 8.0, 1.8 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 26.37 (COCH3), 36.35 (NCH3), 53.08 (CH2), 114.03 (C-4a), 123.73, 124.96 (C-6, C-8), 127.47, 127.89 (C-2′, C-3′), 127.52, 128.79 (C-5, C-4′), 135.23 (C-7), 136.80 (C-1′), 149.03 (C-8a), 153.77 (C-2), 158.78 (C-4), 183.36 (CO); LC-MS (ESI) tR = 12.56 min, 98% purity, m/z [M + H]+ calcd for C18H17N3OS, 324.11; found, 324.1. Anal. calcd for C18H17N3OS: C, 66.85%; H, 5.30%; N, 12.99%. Found: C, 67.15%; H, 4.93%; N, 13.17%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.11 (t, J = 7.4 Hz, 3H, COCH2CH3), 1.18 (t, J = 7.0 Hz, 6H, NCH2CH3), 2.63 (q, J = 7.4 Hz, 2H, COCH2), 3.58 (q, J = 7.0 Hz, 4H, NCH2), 7.11–7.19 (m, 1H, 6-H), 7.25 (dd, J = 8.2, 1.2 Hz, 1H, 8-H), 7.56–7.63 (m, 1H, 7-H), 8.13 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.97 (COCH2CH3), 13.05 (NCH2CH3), 32.04 (COCH2), 43.12 (NCH2), 114.02 (C-4a), 123.34, 124.88 (C-6, C-8), 127.82 (C-5), 135.10 (C-7), 149.36 (C-8a), 152.11 (C-2), 159.06 (C-4), 186.57 (CO); LC-MS (ESI) tR = 11.31 min, 99% purity, m/z [M + H]+ calcd for C15H19N3OS, 290.13; found, 290.1. Anal. calcd for C15H19N3OS: C, 62.25%; H, 6.62%; N, 14.52%. Found: C, 62.41%; H, 6.48%; N, 14.25%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.11 (t, J = 7.4 Hz, 3H, CH3); 2.63 (q, J = 7.3 Hz, 2H, COCH2), 3.62–3.73 (m, 8H, CH2CH2), 7.17–7.25 (m, 1H, 6-H), 7.28 (dd, J = 8.2, 1.3 Hz, 1H, 8-H), 7.60–7.67 (m, 1H, 7-H), 8.14 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.89 (CH3), 31.96 (COCH2), 45.71 (NCH2), 65.75 (OCH2), 114.53 (C-4a), 124.22, 124.98 (C-6, C-8), 127.92 (C-5), 135.22 (C-7), 148.48 (C-8a), 153.66 (C-2), 158.16 (C-4), 186.62 (CO); LC-MS (ESI) (90% H2O to 100% MeCN in 10 min, then 100% MeCN to 20 min, DAD 200–400 nm), tR = 9.54 min, 99% purity, m/z [M + H]+ calcd for C15H17N3O2S, 304.11; found, 304.0. Anal. calcd for C15H17N3O2S: C, 59.39%; H, 5.65%; N, 13.85%. Found: C, 59.38%; H, 5.65%; N, 13.61%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.09 (t, J = 7.4 Hz, 3H, CH2CH3); 2.62 (q, J = 7.4 Hz, 2H, COCH2), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, NCH2), 7.15–7.23 (m, 1H, 6-H), 7.25–7.38 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.59–7.66 (m, 1H, 7-H), 8.16 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.91 (CH2CH3); 32.06 (COCH2), 36.35 (NCH3), 53.07 (NCH2), 114.12 (C-4a), 124.97, 123.71 (C-6, C-8), 127.45, 127.88 (C-2′, C-3′), 127.49, 128.77 (C-5, C-4′), 135.19 (C-7), 136.80 (C-1′), 149.02 (C-8a), 153.82 (C-2), 158.96 (C-4), 186.54 (CO); LC-MS (ESI) (90% H2O to 100% MeCN in 10 min, then 100% MeCN to 20 min, DAD 220–400 nm), tR = 11.32 min, 98% purity, m/z [M + H]+ calcd for C19H19N3OS, 338.13; found, 338.1. Anal. calcd for C19H19N3OS: C, 67.63%; H, 5.68%; N, 12.45%. Found: C, 67.65%; H, 5.56%; N, 12.07%.:1).:1); 1H NMR (600 MHz, DMSO-d6) δ 1.15–1.20 (m, 12H, CHCH3, CH2CH3); 2.80 (hept, J = 6.9 Hz, 1H, CHCH3), 3.58 (q, J = 7.1 Hz, 4H, CH2), 7.13–7.19 (m, 1H, 6-H), 7.25 (dd, J = 8.3, 1.3 Hz, 1H, 8-H), 7.57–7.63 (m, 1H, 7-H), 8.16 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 13.03 (CH2CH3), 18.84 (CHCH3), 37.82 (CHCH3), 43.13 (CH2), 114.14 (C-4a), 123.36, 124.96 (C-6, C-8), 127.85 (C-5), 135.17 (C-7), 149.47 (C-8a), 152.26 (C-2), 160.10 (C-4), 189.17 (CO); LC-MS (ESI) tR = 12.75 min, 96% purity, m/z [M + H]+ calcd for C16H21N3OS, 304.14; found, 304.0. Anal. calcd for C16H21N3OS: C, 63.34%; H, 6.98%; N, 13.85%. Found: C, 63.64%; H, 7.13%; N, 13.45%.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 116–117 °C; Rf = 0.43 (petroleum ether/EtOAc, 10:1); 1H NMR (600 MHz, DMSO-d6) δ 1.17 (d, J = 7.0 Hz, 6H, CH3); 2.81 (hept, J = 7.0 Hz, 1H, CHCH3), 3.66–3.68 (m, 8H, CH2CH2), 7.20–7.26 (m, 1H, 6-H), 7.29 (dd, J = 8.2, 1.3 Hz, 1H, 8-H), 7.61–7.67 (m, 1H, 7-H), 8.18 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 18.80 (CH3), 37.78 (CHCH3), 45.70 (NCH2), 65.75 (OCH2), 114.64 (C-4a), 124.24, 125.06 (C-6, C-8), 127.96 (C-5), 135.31 (C-7), 148.59 (C-8a), 153.85 (C-2), 159.26 (C-4), 189.22 (CO); LC-MS (ESI) tR = 13.07 min, 96% purity, m/z [M + H]+ calcd for C16H19N3O2S, 318.12; found, 318.0. Anal. calcd for C16H19N3O2S: C, 60.55%; H, 6.03%; N, 13.24%. Found: C, 60.42%; H, 6.21%; N, 12.82%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.16 (d, J = 6.9 Hz, 6H, CHCH3), 2.79 (hept, J = 6.9 Hz, 1H, CHCH3), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, CH2), 7.18–7.22 (m, 1H, 6-H), 7.25–7.36 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.62–7.65 (m, 1H, 7-H), 8.20 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 18.81 (CHCH3), 36.41 (NCH3), 37.82 (CHCH3), 53.10 (CH2), 114.25 (C-4a), 123.75, 125.06 (C-6, C-8), 127.45, 127.51 (C-2′, C-3′), 127.92, 128.78 (C-5, C-4′), 135.28 (C-7), 136.81 (C-1′), 149.13 (C-8a), 153.99 (C-2), 159.96 (C-4), 189.17 (CO); LC-MS (ESI) tR = 13.01 min, 97% purity, m/z [M + H]+ calcd for C20H21N3OS, 352.14; found, 352.1. Anal. calcd for C20H21N3OS: C, 68.35%; H, 6.02%; N, 11.96%. Found: C, 68.25%; H, 6.14%; N, 11.64%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 0.95 (d, J = 6.9 Hz, 6H, CHCH3), 1.17 (t, J = 7.0 Hz, 6H, CH2CH3), 2.13 (non, J = 7.0 Hz, 1H, CHCH3), 2.48 (d, J = 6.9 Hz, 2H, CH2CH), 3.58 (q, J = 7.0 Hz, 4H, CH2CH3), 7.13–7.17 (m, 1H, 6-H), 7.24 (dd, J = 8.2, 1.2 Hz, 1H, 8-H), 7.57–7.62 (m, 1H, 7-H), 8.12 (dd, J = 8.1, 1.5 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.02 (CH2CH3), 22.52 (CHCH3), 25.29 (CHCH3), 43.14 (CH2CH3), 47.95 (CH2CH), 114.06 (C-4a), 123.35, 124.88 (C-6, C-8), 127.82 (C-5), 135.12 (C-7), 149.42 (C-8a), 152.10 (C-2), 159.11 (C-4), 185.17 (CO); LC-MS (ESI) tR = 12.97 min, 98% purity, m/z [M + H]+ calcd for C17H23N3OS, 318.16; found, 318.0. HRMS-ESI m/z [M + H]+ calcd for C17H23N3OS, 318.1635; found, 318.1622.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 116–117 °C; Rf = 0.24 (petroleum ether/EtOAc, 10:1); 1H NMR (500 MHz, DMSO-d6) δ 0.96 (d, J = 6.7 Hz, 6H, CHCH3), 2.13 (non, J = 6.8 Hz, 1H, CHCH3), 2.49 (d, J = 6.9 Hz, 2H, CH2CH), 3.65–3.70 (m, 8H, CH2), 7.20–7.24 (m, 1H, 6-H), 7.29 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.62–7.65 (m, 1H, 7-H), 8.15 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 22.54 (CHCH3), 25.30 (CHCH3), 45.70 (NCH2), 47.92 (CH2CH), 65.74 (OCH2), 114.57 (C-4a), 124.23, 125.02 (C-6, C-8), 127.94 (C-5), 135.29 (C-7), 148.58 (C-8a), 153.73 (C-2), 158.52 (C-4), 185.34 (CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 220–500 nm), tR = 12.00 min, 99% purity, m/z [M + H]+ calcd for C17H21N3O2S, 332.14; found, 332.0. Anal. calcd for C17H21N3O2S: C, 61.61%; H, 6.39%; N, 12.68%. Found: C, 61.76%; H, 6.44%; N, 12.43%.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 75–76 °C; Rf = 0.43 (petroleum ether/EtOAc, 10:1); 1H NMR (500 MHz, DMSO-d6) δ 0.93 (d, J = 6.7 Hz, 6H, CHCH3), 2.11 (non, J = 6.7 Hz, 1H, CHCH3), 2.47 (d, J = 7.0 Hz, 2H, CH2CH), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, NCH2), 7.17–7.21 (m, 1H, 6-H), 7.25–7.37 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.60–7.65 (m, 1H, 7-H), 8.15 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 22.51 (CHCH3), 25.28 (CHCH3), 36.42 (NCH3), 47.96 (CH2CH), 53.11 (NCH2), 114.17 (C-4a), 123.75, 124.99 (C-6, C-8), 127.45, 128.79 (C-2′, C-3′), 127.51, 127.91 (C-5, C-4′), 135.25 (C-7), 136.49 (C-1′), 149.09 (C-8a), 153.83 (C-2), 159.06 (C-4), 185.32 (CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 220–500 nm), tR = 13.53 min, 98% purity, m/z [M + H]+ calcd for C21H23N3OS, 366.16; found, 366.0. Anal. calcd for C21H23N3OS: C, 69.01%; H, 6.34%; N, 11.50%. Found: C, 68.72%; H, 6.38%; N, 11.38%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.19 (t, J = 7.1 Hz, 6H, CH3); 3.61 (q, J = 7.0 Hz, 4H, NCH2), 4.68 (s, 2H, CH2Cl), 7.14–7.27 (m, 1H, 6-H), 7.29 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.62–7.70 (m, 1H, 7-H), 8.24 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 12.91 (CH3), 43.29 (NCH2), 46.52 (CH2Cl), 113.93 (C-4a), 123.43, 125.25 (C-6, C-8), 128.01 (C-5), 136.11 (C-7), 150.35 (C-8a), 152.66 (C-2), 165.85 (C-4), 178.10 (CO); LC-MS (ESI) tR = 12.56 min, 98% purity, m/z [M + H]+ calcd for C14H16ClN3OS, 310.07; found, 310.0. Anal. calcd for C14H16ClN3OS: C, 54.27%; H, 5.21%; N, 13.56%. Found: C, 53.97%; H, 5.37%; N, 13.49%.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.66–3.74 (m, 8H, CH2CH2), 4.69 (s, 2H, CH2Cl), 7.22–7.30 (m, 1H, 6-H), 7.33 (dd, J = 8.4, 1.3 Hz, 1H, 8-H), 7.67–7.74 (m, 1H, 7-H), 8.25 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 45.73 (NCH2), 46.44 (CH2Cl), 65.73 (OCH2), 114.43 (C-4a), 124.29, 125.34 (C-6, C-8), 128.09 (C-5), 136.24 (C-7), 149.45 (C-8a), 154.13 (C-2), 164.91 (C-4), 178.24 (CO); LC-MS (ESI) tR = 11.84 min, 99% purity, m/z [M + H]+ calcd for C14H14ClN3O2S, 324.05; found, 324.1. Anal. calcd for C14H14ClN3O2S: C, 51.93%; H, 4.36%; N, 12.98%. Found: C, 51.92%; H, 4.77%; N, 12.92%.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.18 (s, 3H, CH3); 4.68 (s, 2H, CH2Cl), 4.92 (s, 2H, NCH2), 7.21–7.25 (m, 1H, 6-H), 7.26–7.39 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.26–7.39 (m, 1H, 7-H), 8.27 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 36.51 (CH3), 46.54 (CH2Cl), 53.21 (NCH2), 114.06 (C-4a), 123.83, 125.35 (C-6, C-8), 127.48, 128.82 (C-2′, C-3′), 127.57, 128.09 (C-5, C-4′), 136.24 (C-7), 136.64 (C-1′), 150.00 (C-8a), 154.36 (C-2), 165.71 (C-4), 178.14 (CO); LC-MS (ESI) tR = 12.78 min, 97% purity, m/z [M + H]+ calcd for C18H16ClN3OS, 358.07; found, 358.0. Anal. calcd for C18H16ClN3OS: C, 60.41%; H, 4.51%; N, 11.74%. Found: C, 60.38%; H, 4.95%; N, 11.62%.:1).:1); 1H NMR (600 MHz, DMSO-d6) δ 1.17 (t, J = 7.0 Hz, 6H, CH3), 3.58 (q, J = 7.1 Hz, 4H, CH2CH3), 5.26 (s, 2H, OCH2), 7.12–7.18 (m, 1H, 6-H), 7.26 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.32–7.48 (m, 5H, 2′′-H, 3′′-H, 4′′-H), 7.59–7.65 (m, 1H, 7-H), 8.16 (dd, J = 8.3, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 12.98 (CH3), 43.21 (CH2CH3), 67.79 (OCH2), 113.90 (C-4a), 123.48, 124.89 (C-6, C-8), 127.90, 128.39 (C-5, C-4′′), 128.53, 128.60 (C-2′′, C-3′′), 135.66 (C-7), 136.06 (C-1′′), 149.54 (C-8a), 152.25 (C-2), 160.74, 166.24 (C-4, CO); LC-MS (ESI) tR = 13.54 min, 99% purity, m/z [M + H]+ calcd for C20H21N3O2S, 368.14; found, 368.0. Anal. calcd for C20H21N3O2S: C, 65.37%; H, 5.76%; N, 11.44%. Found: C, 65.38%; H, 5.86%; N, 10.97%.:1); 1H NMR (600 MHz, DMSO-d6) δ 3.63–372 (m, 8H, CH2CH2), 5.26 (s, 2H, CO2CH2), 7.20–7.23 (m, 1H, 6-H), 7.29–7.46 (m, 6H, 8-H, 2′′-H, 3′′-H, 4′′-H), 7.64–7.68 (m, 1H, 7-H), 8.18 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 45.69 (NCH2), 65.67 (OCH2CH2), 67.86 (CO2CH2), 114.39 (C-4a), 124.30, 124.97 (C-6, C-8), 127.96, 128.39 (C-5, C-4′′), 128.54, 128.59 (C-2′′, C-3′′), 135.77 (C-7), 135.91 (C-1′′), 148.66 (C-8a), 153.79 (C-2), 160.69, 165.57 (C-4, CO); LC-MS (ESI) tR = 10.47 min, 99% purity, m/z [M + H]+ calcd for C20H19N3O3S, 382.12; found, 382.2. HRMS-ESI m/z [M + H]+ calcd for C20H19N3O3S, 382.1220; found, 382.1210.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.14 (s, 3H, CH3), 4.88 (s, 2H, NCH2), 5.24 (s, 2H, OCH2), 7.17–7.21 (m, 1H, 6-H), 7.26–7.44 (m, 11H, 8-H, 2′-H, 3′-H, 4′-H, 2′′-H, 3′′-H, 4′′-H), 7.63–7.67 (m, 1H, 7-H), 8.19 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 36.43 (NCH3), 53.15 (NCH2), 67.85 (OCH2), 114.04 (C-4a), 123.88, 125.02 (C-6, C-8), 127.47, 128.51, 128.60, 128.82 (C-2′, C-3′, C2′′, C3′′), 127.56, 127.99, 128.39 (C-5, C-4′, C-4′′), 135.80, 135.99, 136.68 (C-7, C-1′, C-1′′), 149.22 (C-8a), 154.00 (C-2), 160.75, 166.24 (C-4, CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 200–500 nm), tR = 13.81 min, 99% purity, m/z [M + H]+ calcd for C24H21N3O2S, 416.14; found, 416.1. Anal. calcd for C24H21N3O2S: C, 69.38%; H, 5.09%; N, 10.11%. Found: C, 68.96%; H, 5.50%; N, 10.02%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.18 (t, J = 7.1 Hz, 6H, CH3), 3.49 (q, J = 7.1 Hz, 4H, NCH2), 7.12–7.15 (m, 1H, 6-H), 7.18 (d, J = 8.2 Hz, 1H, 8-H), 7.63–7.67 (m, 1H, 7-H), 7.86 (dd, J = 7.8, 1.5 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.29 (CH3), 41.90 (CH2), 111.79 (C-4a), 122.82, 123.97 (C-6, C-8), 128.14 (C-5), 136.86 (C-7), 151.06, 153.31 (C-2, C-8a), 159.48 (C-4); LC-MS (ESI) tR = 12.89 min, 99% purity, m/z [M + H]+ calcd for C12H14N2O2, 219.11; found, 219.0.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.60–3.68 (m, 8H, CH2), 7.18–7.22 (m, 2H, 6-H, 8-H), 7.67–7.71 (m, 1H, 7-H), 7.89 (dd, J = 7.7, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 44.26 (NCH2), 65.63 (OCH2), 112.26 (C-4a), 123.56, 124.16 (C-6, C-8), 128.30 (C-5), 137.06 (C-7), 150.38, 153.32 (C-8a, C-2), 159.18 (C-4); LC-MS (ESI) tR = 7.31 min, 99% purity, m/z [M + H]+ calcd for C12H12N2O3, 233.09; found, 233.2.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.05 (s, 3H, CH3), 4.73 (s, 2H, CH2), 7.16–7.19 (m, 1H, 6-H), 7.21–7.37 (m, 6H, 2′-H, 3′-H, 4′-H, 8-H), 7.66–7.69 (m, 1H, 7-H), 7.89 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 35.13 (CH3), 51.92 (CH2), 112.03 (C-4a), 123.25, 124.15 (C-6, C-8), 127.57, 128.29, 128.80 (C-5, C-2′, C-3′, C-4′), 136.95, 137.04 (C-7, C-1′), 150.78, 154.23 (C-2, C-8a), 159.38 (C-4); LC-MS (ESI) tR = 9.55 min, 99% purity, m/z [M + H]+ calcd for C16H14N2O2, 267.11; found, 267.0.:500 with assay buffer containing 5 mM DTT and incubated for 30 min at 37 °C. Inhibitor stock solutions were prepared in DMSO. A 10 mM stock solution of the chromogenic substrate Z-Arg-Arg-pNA was prepared with DMSO. The final concentration of DMSO was 2%, and the final concentration of the substrate was 500 μM (0.45 Km).2 Assays were performed with a final concentration of 216 ng mL−1 or 288 ng mL−1 of cathepsin B. Into a cuvette containing 920 μL or 900 μL assay buffer, inhibitor solution and DMSO in a total volume of 15 μL, and 5 μL of the substrate solution were added and thoroughly mixed. The reaction was initiated by adding 60 μL or 80 μL of the cathepsin B solution.:100 with assay buffer containing 5 mM DTT and incubated for 30 min at 37 °C. Inhibitor stock solutions were prepared in DMSO. A 10 mM stock solution of the chromogenic substrate Z-Phe-Arg-pNA was prepared with DMSO. The final concentration of DMSO was 2%, and the final concentration of the substrate was 100 μM (5.88 Km).2 Assays were performed with a final concentration of 54 ng mL−1 of cathepsin L. Into a cuvette containing 940 μL assay buffer, inhibitor solution and DMSO in a total volume of 10 μL, and 10 μL of the substrate solution were added and thoroughly mixed. The reaction was initiated by adding 40 μL of the cathepsin L solution.
:10 up to 0:100 in 9 min, 0:100 for 5 min. Elemental analyses were performed with a Vario EL apparatus for C, H, and N. The purity of the compounds was determined by HPLC-UV obtained on an LC-MS instrument (Applied Biosystems API 2000 LC/MS/MS, HPLC Agilent 1100) using the procedure as follows: dissolving of the compounds at a concentration of 1.0 mg mL−1 in MeCN and if necessary sonicated to complete dissolving. Then, 10 μL of the substance solution was injected into a Phenomenex Luna C18 HPLC column (50 × 2.00 mm, particle size 3 μm) and elution performed with a gradient of water/MeOH either containing 2 mM ammonium acetate from 90:10 up to 0:100 for 30 min at a flow rate of 300 μL min−1, starting the gradient after 1 min. UV absorption was detected from 220 to 400 nm using a diode array detector, unless noted otherwise.
:1); 1H NMR (500 MHz, DMSO-d6) δ 7.15–7.61 (m, 7H, 3-H, 5-H, 2′-H, 3′-H, 4′-H), 7.68–7.75 (m, 1H, 4-H), 7.84 (dd, J = 7.8, 1.5 Hz, 1H, 6-H), 10.32 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 108.04 (C-1), 116.87 (CN), 121.83, 125.72, 125.76, 126.19, 129.60 (C-3, C-5, C-2′, C-3′, C-4′), 133.52, 134.10 (C-4, C-6), 140.09, 150.67, 152.51 (C-1′, C-2, CO).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.12 (t, J = 7.0 Hz, 6H, CH3), 3.34 (q, J = 7.1 Hz, 4H, CH2), 7.21–7.46 (m, 2H, 3-H, 5-H), 7.58–7.61 (m, 1H, 4-H), 7.70 (dd, J = 7.8, 1.6 Hz, 1H, 6-H), 8.44 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 13.89 (CH3), 40.89 (CH2), 108.22 (C-1), 117.46 (CN), 124.30, 125.59 (C-3, C-5), 132.71, 133.30 (C-4, C-6), 142.98 (C-2), 154.42 (CO); LC-MS (ESI) tR = 6.54 min, 99% purity, m/z [M + H]+ calcd for C12H15N3O, 218.28; found, 218.3. HRMS-ESI m/z [M + H]+ calcd for C12H15N3O, 218.1288; found, 218.1295.:2); 1H NMR (600 MHz, DMSO-d6) δ 3.42–3.47 (m, 4H, NCH2), 3.59–3.64 (m, 4H, OCH2), 7.24–7.42 (m, 2H, 3-H, 5-H), 7.60–7.63 (m, 1H, 4-H), 7.73 (dd, J = 7.7, 1.6, Hz, 1H, 6-H), 8.86 (s, 1H, NH); 13C NMR (151 MHz, DMSO-d6) δ 44.57 (NCH2), 66.10 (OCH2), 108.34 (C-1), 117.40 (CN), 124.66, 125.46 (C-3, C-5), 132.97, 133.53 (C-4, C-6), 142.63 (C-2), 155.23 (CO); LC-MS (ESI) tR = 3.89 min, 99% purity, m/z [M + H]+ calcd for C12H13N3O2, 232.10; found, 232.2. HRMS-ESI m/z [M + H]+ calcd for C12H13N3O2, 232.1081; found, 232.1074.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.92 (s, 3H, CH3), 4.56 (s, 2H, CH2), 7.24–7.48 (m, 7H, 3-H, 5-H, 2′-H, 3′-H, 4′-H), 7.58–7.66 (m, 1H, 4-H), 7.73 (dd, J = 7.7, 1.7 Hz, 1H, 6-H), 8.73 (s, 1H, NH); 13C NMR (126 MHz, DMSO-d6) δ 34.26 (CH3), 51.41 (CH2), 108.54 (C-1), 117.49 (CN), 124.59, 125.50 (C-3, C-5), 127.18, 127.47, 128.57 (C-2′, C-3′, C-4′), 132.90, 133.45 (C-4, C-6), 138.10 (C-1′), 142.78 (C-2), 155.74 (CO); LC-MS (ESI) tR = 7.84 min, 99% purity, m/z [M + H]+ calcd for C16H15N3O, 266.32; found, 266.3. HRMS-ESI m/z [M + H]+ calcd for C16H15N3O, 266.1288; found, 266.1282.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.18 (t, J = 7.0 Hz, 6H, CH2CH3), 2.32 (s, 3H, COCH3), 3.58 (q, J = 7.1 Hz, 4H, CH2), 7.13–7.17 (m, 1H, 6-H), 7.25 (dd, J = 8.2, 1.1 Hz, 1H, 8-H), 7.58–7.60 (m, 1H, 7-H), 8.11 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.04 (CH2CH3), 26.31 (COCH3), 43.13 (CH2), 113.92 (C-4a), 123.34, 124.84 (C-6, C-8), 127.81 (C-5), 135.10 (C-7), 149.34 (C-8a), 152.02 (C-2), 158.72 (C-4), 183.39 (CO); LC-MS (ESI) tR = 13.77 min, 97% purity, m/z [M + H]+ calcd for C14H17N3OS, 276.11; found, 276.1. HRMS-ESI m/z [M + H]+ calcd for C14H17N3OS, 276.1165; found, 276.1157.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3), 3.61–3.73 (m, 8H, CH2), 7.18–7.25 (m, 1H, 6-H), 7.28 (dd, J = 8.4, 1.2 Hz, 1H, 8-H), 7.60–7.68 (m, 1H, 7-H), 8.13 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 26.26 (CH3), 45.69 (NCH2), 65.72 (OCH2), 114.41 (C-4a), 124.18, 124.93 (C-6, C-8), 127.89 (C-5), 135.21 (C-7), 148.46 (C-8a), 153.58 (C-2), 157.99 (C-4), 183.38 (CO); LC-MS (ESI) tR = 11.45 min, 98% purity, m/z [M + H]+ calcd for C14H15N3O2S, 290.09; found, 290.1. Anal. calcd for C14H15N3O2S: C, 58.11%; H, 5.23%; N, 14.52%. Found: C, 58.01%; H, 5.32%; N, 14.37%.:1); 1H NMR (500 MHz, DMSO-d6) δ 2.31 (s, 3H, COCH3); 3.14 (s, 3H, NCH3), 4.88 (s, 2H, CH2), 7.15–7.23 (m, 1H, 6-H), 7.23–7.40 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.59–7.66 (m, 1H, 7-H), 8.15 (dd, J = 8.0, 1.8 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 26.37 (COCH3), 36.35 (NCH3), 53.08 (CH2), 114.03 (C-4a), 123.73, 124.96 (C-6, C-8), 127.47, 127.89 (C-2′, C-3′), 127.52, 128.79 (C-5, C-4′), 135.23 (C-7), 136.80 (C-1′), 149.03 (C-8a), 153.77 (C-2), 158.78 (C-4), 183.36 (CO); LC-MS (ESI) tR = 12.56 min, 98% purity, m/z [M + H]+ calcd for C18H17N3OS, 324.11; found, 324.1. Anal. calcd for C18H17N3OS: C, 66.85%; H, 5.30%; N, 12.99%. Found: C, 67.15%; H, 4.93%; N, 13.17%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.11 (t, J = 7.4 Hz, 3H, COCH2CH3), 1.18 (t, J = 7.0 Hz, 6H, NCH2CH3), 2.63 (q, J = 7.4 Hz, 2H, COCH2), 3.58 (q, J = 7.0 Hz, 4H, NCH2), 7.11–7.19 (m, 1H, 6-H), 7.25 (dd, J = 8.2, 1.2 Hz, 1H, 8-H), 7.56–7.63 (m, 1H, 7-H), 8.13 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.97 (COCH2CH3), 13.05 (NCH2CH3), 32.04 (COCH2), 43.12 (NCH2), 114.02 (C-4a), 123.34, 124.88 (C-6, C-8), 127.82 (C-5), 135.10 (C-7), 149.36 (C-8a), 152.11 (C-2), 159.06 (C-4), 186.57 (CO); LC-MS (ESI) tR = 11.31 min, 99% purity, m/z [M + H]+ calcd for C15H19N3OS, 290.13; found, 290.1. Anal. calcd for C15H19N3OS: C, 62.25%; H, 6.62%; N, 14.52%. Found: C, 62.41%; H, 6.48%; N, 14.25%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.11 (t, J = 7.4 Hz, 3H, CH3); 2.63 (q, J = 7.3 Hz, 2H, COCH2), 3.62–3.73 (m, 8H, CH2CH2), 7.17–7.25 (m, 1H, 6-H), 7.28 (dd, J = 8.2, 1.3 Hz, 1H, 8-H), 7.60–7.67 (m, 1H, 7-H), 8.14 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.89 (CH3), 31.96 (COCH2), 45.71 (NCH2), 65.75 (OCH2), 114.53 (C-4a), 124.22, 124.98 (C-6, C-8), 127.92 (C-5), 135.22 (C-7), 148.48 (C-8a), 153.66 (C-2), 158.16 (C-4), 186.62 (CO); LC-MS (ESI) (90% H2O to 100% MeCN in 10 min, then 100% MeCN to 20 min, DAD 200–400 nm), tR = 9.54 min, 99% purity, m/z [M + H]+ calcd for C15H17N3O2S, 304.11; found, 304.0. Anal. calcd for C15H17N3O2S: C, 59.39%; H, 5.65%; N, 13.85%. Found: C, 59.38%; H, 5.65%; N, 13.61%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.09 (t, J = 7.4 Hz, 3H, CH2CH3); 2.62 (q, J = 7.4 Hz, 2H, COCH2), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, NCH2), 7.15–7.23 (m, 1H, 6-H), 7.25–7.38 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.59–7.66 (m, 1H, 7-H), 8.16 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 8.91 (CH2CH3); 32.06 (COCH2), 36.35 (NCH3), 53.07 (NCH2), 114.12 (C-4a), 124.97, 123.71 (C-6, C-8), 127.45, 127.88 (C-2′, C-3′), 127.49, 128.77 (C-5, C-4′), 135.19 (C-7), 136.80 (C-1′), 149.02 (C-8a), 153.82 (C-2), 158.96 (C-4), 186.54 (CO); LC-MS (ESI) (90% H2O to 100% MeCN in 10 min, then 100% MeCN to 20 min, DAD 220–400 nm), tR = 11.32 min, 98% purity, m/z [M + H]+ calcd for C19H19N3OS, 338.13; found, 338.1. Anal. calcd for C19H19N3OS: C, 67.63%; H, 5.68%; N, 12.45%. Found: C, 67.65%; H, 5.56%; N, 12.07%.:1).:1); 1H NMR (600 MHz, DMSO-d6) δ 1.15–1.20 (m, 12H, CHCH3, CH2CH3); 2.80 (hept, J = 6.9 Hz, 1H, CHCH3), 3.58 (q, J = 7.1 Hz, 4H, CH2), 7.13–7.19 (m, 1H, 6-H), 7.25 (dd, J = 8.3, 1.3 Hz, 1H, 8-H), 7.57–7.63 (m, 1H, 7-H), 8.16 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 13.03 (CH2CH3), 18.84 (CHCH3), 37.82 (CHCH3), 43.13 (CH2), 114.14 (C-4a), 123.36, 124.96 (C-6, C-8), 127.85 (C-5), 135.17 (C-7), 149.47 (C-8a), 152.26 (C-2), 160.10 (C-4), 189.17 (CO); LC-MS (ESI) tR = 12.75 min, 96% purity, m/z [M + H]+ calcd for C16H21N3OS, 304.14; found, 304.0. Anal. calcd for C16H21N3OS: C, 63.34%; H, 6.98%; N, 13.85%. Found: C, 63.64%; H, 7.13%; N, 13.45%.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 116–117 °C; Rf = 0.43 (petroleum ether/EtOAc, 10:1); 1H NMR (600 MHz, DMSO-d6) δ 1.17 (d, J = 7.0 Hz, 6H, CH3); 2.81 (hept, J = 7.0 Hz, 1H, CHCH3), 3.66–3.68 (m, 8H, CH2CH2), 7.20–7.26 (m, 1H, 6-H), 7.29 (dd, J = 8.2, 1.3 Hz, 1H, 8-H), 7.61–7.67 (m, 1H, 7-H), 8.18 (dd, J = 8.1, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 18.80 (CH3), 37.78 (CHCH3), 45.70 (NCH2), 65.75 (OCH2), 114.64 (C-4a), 124.24, 125.06 (C-6, C-8), 127.96 (C-5), 135.31 (C-7), 148.59 (C-8a), 153.85 (C-2), 159.26 (C-4), 189.22 (CO); LC-MS (ESI) tR = 13.07 min, 96% purity, m/z [M + H]+ calcd for C16H19N3O2S, 318.12; found, 318.0. Anal. calcd for C16H19N3O2S: C, 60.55%; H, 6.03%; N, 13.24%. Found: C, 60.42%; H, 6.21%; N, 12.82%.:1); 1H NMR (500 MHz, DMSO-d6) δ 1.16 (d, J = 6.9 Hz, 6H, CHCH3), 2.79 (hept, J = 6.9 Hz, 1H, CHCH3), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, CH2), 7.18–7.22 (m, 1H, 6-H), 7.25–7.36 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.62–7.65 (m, 1H, 7-H), 8.20 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 18.81 (CHCH3), 36.41 (NCH3), 37.82 (CHCH3), 53.10 (CH2), 114.25 (C-4a), 123.75, 125.06 (C-6, C-8), 127.45, 127.51 (C-2′, C-3′), 127.92, 128.78 (C-5, C-4′), 135.28 (C-7), 136.81 (C-1′), 149.13 (C-8a), 153.99 (C-2), 159.96 (C-4), 189.17 (CO); LC-MS (ESI) tR = 13.01 min, 97% purity, m/z [M + H]+ calcd for C20H21N3OS, 352.14; found, 352.1. Anal. calcd for C20H21N3OS: C, 68.35%; H, 6.02%; N, 11.96%. Found: C, 68.25%; H, 6.14%; N, 11.64%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 0.95 (d, J = 6.9 Hz, 6H, CHCH3), 1.17 (t, J = 7.0 Hz, 6H, CH2CH3), 2.13 (non, J = 7.0 Hz, 1H, CHCH3), 2.48 (d, J = 6.9 Hz, 2H, CH2CH), 3.58 (q, J = 7.0 Hz, 4H, CH2CH3), 7.13–7.17 (m, 1H, 6-H), 7.24 (dd, J = 8.2, 1.2 Hz, 1H, 8-H), 7.57–7.62 (m, 1H, 7-H), 8.12 (dd, J = 8.1, 1.5 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.02 (CH2CH3), 22.52 (CHCH3), 25.29 (CHCH3), 43.14 (CH2CH3), 47.95 (CH2CH), 114.06 (C-4a), 123.35, 124.88 (C-6, C-8), 127.82 (C-5), 135.12 (C-7), 149.42 (C-8a), 152.10 (C-2), 159.11 (C-4), 185.17 (CO); LC-MS (ESI) tR = 12.97 min, 98% purity, m/z [M + H]+ calcd for C17H23N3OS, 318.16; found, 318.0. HRMS-ESI m/z [M + H]+ calcd for C17H23N3OS, 318.1635; found, 318.1622.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 116–117 °C; Rf = 0.24 (petroleum ether/EtOAc, 10:1); 1H NMR (500 MHz, DMSO-d6) δ 0.96 (d, J = 6.7 Hz, 6H, CHCH3), 2.13 (non, J = 6.8 Hz, 1H, CHCH3), 2.49 (d, J = 6.9 Hz, 2H, CH2CH), 3.65–3.70 (m, 8H, CH2), 7.20–7.24 (m, 1H, 6-H), 7.29 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.62–7.65 (m, 1H, 7-H), 8.15 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 22.54 (CHCH3), 25.30 (CHCH3), 45.70 (NCH2), 47.92 (CH2CH), 65.74 (OCH2), 114.57 (C-4a), 124.23, 125.02 (C-6, C-8), 127.94 (C-5), 135.29 (C-7), 148.58 (C-8a), 153.73 (C-2), 158.52 (C-4), 185.34 (CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 220–500 nm), tR = 12.00 min, 99% purity, m/z [M + H]+ calcd for C17H21N3O2S, 332.14; found, 332.0. Anal. calcd for C17H21N3O2S: C, 61.61%; H, 6.39%; N, 12.68%. Found: C, 61.76%; H, 6.44%; N, 12.43%.:1. This was kept at −18 °C for 7 d and the precipitate was collected by suction filtration: mp 75–76 °C; Rf = 0.43 (petroleum ether/EtOAc, 10:1); 1H NMR (500 MHz, DMSO-d6) δ 0.93 (d, J = 6.7 Hz, 6H, CHCH3), 2.11 (non, J = 6.7 Hz, 1H, CHCH3), 2.47 (d, J = 7.0 Hz, 2H, CH2CH), 3.14 (s, 3H, NCH3), 4.88 (s, 2H, NCH2), 7.17–7.21 (m, 1H, 6-H), 7.25–7.37 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.60–7.65 (m, 1H, 7-H), 8.15 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 22.51 (CHCH3), 25.28 (CHCH3), 36.42 (NCH3), 47.96 (CH2CH), 53.11 (NCH2), 114.17 (C-4a), 123.75, 124.99 (C-6, C-8), 127.45, 128.79 (C-2′, C-3′), 127.51, 127.91 (C-5, C-4′), 135.25 (C-7), 136.49 (C-1′), 149.09 (C-8a), 153.83 (C-2), 159.06 (C-4), 185.32 (CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 220–500 nm), tR = 13.53 min, 98% purity, m/z [M + H]+ calcd for C21H23N3OS, 366.16; found, 366.0. Anal. calcd for C21H23N3OS: C, 69.01%; H, 6.34%; N, 11.50%. Found: C, 68.72%; H, 6.38%; N, 11.38%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.19 (t, J = 7.1 Hz, 6H, CH3); 3.61 (q, J = 7.0 Hz, 4H, NCH2), 4.68 (s, 2H, CH2Cl), 7.14–7.27 (m, 1H, 6-H), 7.29 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.62–7.70 (m, 1H, 7-H), 8.24 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 12.91 (CH3), 43.29 (NCH2), 46.52 (CH2Cl), 113.93 (C-4a), 123.43, 125.25 (C-6, C-8), 128.01 (C-5), 136.11 (C-7), 150.35 (C-8a), 152.66 (C-2), 165.85 (C-4), 178.10 (CO); LC-MS (ESI) tR = 12.56 min, 98% purity, m/z [M + H]+ calcd for C14H16ClN3OS, 310.07; found, 310.0. Anal. calcd for C14H16ClN3OS: C, 54.27%; H, 5.21%; N, 13.56%. Found: C, 53.97%; H, 5.37%; N, 13.49%.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.66–3.74 (m, 8H, CH2CH2), 4.69 (s, 2H, CH2Cl), 7.22–7.30 (m, 1H, 6-H), 7.33 (dd, J = 8.4, 1.3 Hz, 1H, 8-H), 7.67–7.74 (m, 1H, 7-H), 8.25 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 45.73 (NCH2), 46.44 (CH2Cl), 65.73 (OCH2), 114.43 (C-4a), 124.29, 125.34 (C-6, C-8), 128.09 (C-5), 136.24 (C-7), 149.45 (C-8a), 154.13 (C-2), 164.91 (C-4), 178.24 (CO); LC-MS (ESI) tR = 11.84 min, 99% purity, m/z [M + H]+ calcd for C14H14ClN3O2S, 324.05; found, 324.1. Anal. calcd for C14H14ClN3O2S: C, 51.93%; H, 4.36%; N, 12.98%. Found: C, 51.92%; H, 4.77%; N, 12.92%.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.18 (s, 3H, CH3); 4.68 (s, 2H, CH2Cl), 4.92 (s, 2H, NCH2), 7.21–7.25 (m, 1H, 6-H), 7.26–7.39 (m, 6H, 8-H, 2′-H, 3′-H, 4′-H), 7.26–7.39 (m, 1H, 7-H), 8.27 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 36.51 (CH3), 46.54 (CH2Cl), 53.21 (NCH2), 114.06 (C-4a), 123.83, 125.35 (C-6, C-8), 127.48, 128.82 (C-2′, C-3′), 127.57, 128.09 (C-5, C-4′), 136.24 (C-7), 136.64 (C-1′), 150.00 (C-8a), 154.36 (C-2), 165.71 (C-4), 178.14 (CO); LC-MS (ESI) tR = 12.78 min, 97% purity, m/z [M + H]+ calcd for C18H16ClN3OS, 358.07; found, 358.0. Anal. calcd for C18H16ClN3OS: C, 60.41%; H, 4.51%; N, 11.74%. Found: C, 60.38%; H, 4.95%; N, 11.62%.:1).:1); 1H NMR (600 MHz, DMSO-d6) δ 1.17 (t, J = 7.0 Hz, 6H, CH3), 3.58 (q, J = 7.1 Hz, 4H, CH2CH3), 5.26 (s, 2H, OCH2), 7.12–7.18 (m, 1H, 6-H), 7.26 (dd, J = 8.3, 1.2 Hz, 1H, 8-H), 7.32–7.48 (m, 5H, 2′′-H, 3′′-H, 4′′-H), 7.59–7.65 (m, 1H, 7-H), 8.16 (dd, J = 8.3, 1.6 Hz, 1H, 5-H); 13C NMR (151 MHz, DMSO-d6) δ 12.98 (CH3), 43.21 (CH2CH3), 67.79 (OCH2), 113.90 (C-4a), 123.48, 124.89 (C-6, C-8), 127.90, 128.39 (C-5, C-4′′), 128.53, 128.60 (C-2′′, C-3′′), 135.66 (C-7), 136.06 (C-1′′), 149.54 (C-8a), 152.25 (C-2), 160.74, 166.24 (C-4, CO); LC-MS (ESI) tR = 13.54 min, 99% purity, m/z [M + H]+ calcd for C20H21N3O2S, 368.14; found, 368.0. Anal. calcd for C20H21N3O2S: C, 65.37%; H, 5.76%; N, 11.44%. Found: C, 65.38%; H, 5.86%; N, 10.97%.:1); 1H NMR (600 MHz, DMSO-d6) δ 3.63–372 (m, 8H, CH2CH2), 5.26 (s, 2H, CO2CH2), 7.20–7.23 (m, 1H, 6-H), 7.29–7.46 (m, 6H, 8-H, 2′′-H, 3′′-H, 4′′-H), 7.64–7.68 (m, 1H, 7-H), 8.18 (dd, J = 8.2, 1.7 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 45.69 (NCH2), 65.67 (OCH2CH2), 67.86 (CO2CH2), 114.39 (C-4a), 124.30, 124.97 (C-6, C-8), 127.96, 128.39 (C-5, C-4′′), 128.54, 128.59 (C-2′′, C-3′′), 135.77 (C-7), 135.91 (C-1′′), 148.66 (C-8a), 153.79 (C-2), 160.69, 165.57 (C-4, CO); LC-MS (ESI) tR = 10.47 min, 99% purity, m/z [M + H]+ calcd for C20H19N3O3S, 382.12; found, 382.2. HRMS-ESI m/z [M + H]+ calcd for C20H19N3O3S, 382.1220; found, 382.1210.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.14 (s, 3H, CH3), 4.88 (s, 2H, NCH2), 5.24 (s, 2H, OCH2), 7.17–7.21 (m, 1H, 6-H), 7.26–7.44 (m, 11H, 8-H, 2′-H, 3′-H, 4′-H, 2′′-H, 3′′-H, 4′′-H), 7.63–7.67 (m, 1H, 7-H), 8.19 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 36.43 (NCH3), 53.15 (NCH2), 67.85 (OCH2), 114.04 (C-4a), 123.88, 125.02 (C-6, C-8), 127.47, 128.51, 128.60, 128.82 (C-2′, C-3′, C2′′, C3′′), 127.56, 127.99, 128.39 (C-5, C-4′, C-4′′), 135.80, 135.99, 136.68 (C-7, C-1′, C-1′′), 149.22 (C-8a), 154.00 (C-2), 160.75, 166.24 (C-4, CO); LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH to 20 min, DAD 200–500 nm), tR = 13.81 min, 99% purity, m/z [M + H]+ calcd for C24H21N3O2S, 416.14; found, 416.1. Anal. calcd for C24H21N3O2S: C, 69.38%; H, 5.09%; N, 10.11%. Found: C, 68.96%; H, 5.50%; N, 10.02%.:1).:1); 1H NMR (500 MHz, DMSO-d6) δ 1.18 (t, J = 7.1 Hz, 6H, CH3), 3.49 (q, J = 7.1 Hz, 4H, NCH2), 7.12–7.15 (m, 1H, 6-H), 7.18 (d, J = 8.2 Hz, 1H, 8-H), 7.63–7.67 (m, 1H, 7-H), 7.86 (dd, J = 7.8, 1.5 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 13.29 (CH3), 41.90 (CH2), 111.79 (C-4a), 122.82, 123.97 (C-6, C-8), 128.14 (C-5), 136.86 (C-7), 151.06, 153.31 (C-2, C-8a), 159.48 (C-4); LC-MS (ESI) tR = 12.89 min, 99% purity, m/z [M + H]+ calcd for C12H14N2O2, 219.11; found, 219.0.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.60–3.68 (m, 8H, CH2), 7.18–7.22 (m, 2H, 6-H, 8-H), 7.67–7.71 (m, 1H, 7-H), 7.89 (dd, J = 7.7, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 44.26 (NCH2), 65.63 (OCH2), 112.26 (C-4a), 123.56, 124.16 (C-6, C-8), 128.30 (C-5), 137.06 (C-7), 150.38, 153.32 (C-8a, C-2), 159.18 (C-4); LC-MS (ESI) tR = 7.31 min, 99% purity, m/z [M + H]+ calcd for C12H12N2O3, 233.09; found, 233.2.:1); 1H NMR (500 MHz, DMSO-d6) δ 3.05 (s, 3H, CH3), 4.73 (s, 2H, CH2), 7.16–7.19 (m, 1H, 6-H), 7.21–7.37 (m, 6H, 2′-H, 3′-H, 4′-H, 8-H), 7.66–7.69 (m, 1H, 7-H), 7.89 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C NMR (126 MHz, DMSO-d6) δ 35.13 (CH3), 51.92 (CH2), 112.03 (C-4a), 123.25, 124.15 (C-6, C-8), 127.57, 128.29, 128.80 (C-5, C-2′, C-3′, C-4′), 136.95, 137.04 (C-7, C-1′), 150.78, 154.23 (C-2, C-8a), 159.38 (C-4); LC-MS (ESI) tR = 9.55 min, 99% purity, m/z [M + H]+ calcd for C16H14N2O2, 267.11; found, 267.0.:500 with assay buffer containing 5 mM DTT and incubated for 30 min at 37 °C. Inhibitor stock solutions were prepared in DMSO. A 10 mM stock solution of the chromogenic substrate Z-Arg-Arg-pNA was prepared with DMSO. The final concentration of DMSO was 2%, and the final concentration of the substrate was 500 μM (0.45 Km).2 Assays were performed with a final concentration of 216 ng mL−1 or 288 ng mL−1 of cathepsin B. Into a cuvette containing 920 μL or 900 μL assay buffer, inhibitor solution and DMSO in a total volume of 15 μL, and 5 μL of the substrate solution were added and thoroughly mixed. The reaction was initiated by adding 60 μL or 80 μL of the cathepsin B solution.:100 with assay buffer containing 5 mM DTT and incubated for 30 min at 37 °C. Inhibitor stock solutions were prepared in DMSO. A 10 mM stock solution of the chromogenic substrate Z-Phe-Arg-pNA was prepared with DMSO. The final concentration of DMSO was 2%, and the final concentration of the substrate was 100 μM (5.88 Km).2 Assays were performed with a final concentration of 54 ng mL−1 of cathepsin L. Into a cuvette containing 940 μL assay buffer, inhibitor solution and DMSO in a total volume of 10 μL, and 10 μL of the substrate solution were added and thoroughly mixed. The reaction was initiated by adding 40 μL of the cathepsin L solution.Acknowledgements
G. S. thanks Prof. A. C. Filippou for support. We thank Andrea Wiesel for performing the cathepsin B assay, Marion Schneider for LC/MS and HRMS measurements, Sabine Terhart-Krabbe and Annette Reiner for recording NMR spectra.Notes and references
- (a) J. Y. Gauthier, N. Chauret, W. Cromlish, S. Desmarais, L. T. Duong, J. P. Falgueyret, D. B. Kimmel, S. Lamontagne, S. Léger, T. LeRiche, C. S. Li, F. Massé, D. J. McKay, D. A. Nicoll-Griffith, R. M. Oballa, J. T. Palmer, M. D. Percival, D. Riendeau, J. Robichaud, G. A. Rodan, S. B. Rodan, C. Seto, M. Thérien, V. L. Truong, M. C. Venuti, G. Wesolowski, R. N. Young, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2008, 18, 923 CrossRef CAS PubMed; (b) R. Löser, M. Frizler, K. Schilling and M. Gütschow, Angew. Chem., Int. Ed., 2008, 47, 4331 CrossRef PubMed; (c) W. C. Black, Curr. Top. Med. Chem., 2010, 10, 745 CrossRef CAS PubMed; (d) M. Frizler, F. Lohr, M. Lülsdorff and M. Gütschow, Chem.–Eur. J., 2011, 17, 11419 CrossRef CAS PubMed.
- M. Frizler, F. Lohr, N. Furtmann, J. Kläs and M. Gütschow, J. Med. Chem., 2011, 54, 396 CrossRef CAS PubMed.
- R. G. Russell, Curr. Opin. Pharmacol., 2015, 22, 115 CrossRef CAS PubMed.
- (a) J. Liebscher and E. Mitzner, Synthesis, 1985, 414 CrossRef CAS; (b) K. Burger, E. Huber, T. Kahl, H. Partscht and M. Ganzer, Synthesis, 1988, 44 CrossRef CAS; (c) T. Takido, M. Toriyama and K. Itabashi, Synthesis, 1988, 404 CrossRef CAS; (d) H. Matsumoto, S. Hara, N. Nagata, K. Ikeda and Y. Mizuno, Heterocycles, 1995, 41, 47 CrossRef CAS; (e) M. Gütschow and J. C. Powers, J. Org. Chem., 2001, 66, 4723 CrossRef; (f) A. Robin, K. Julienne, J. C. Meslin and D. Deniaud, Tetrahedron Lett., 2004, 45, 9557 CrossRef CAS; (g) G. W. Wang, J. X. Li and Y. Xu, Org. Biomol. Chem., 2008, 6, 2995 RSC.
- M. Gütschow, S. Leistner and M. Pink, J. Heterocycl. Chem., 1992, 29, 279 CrossRef.
- For modern utilizations of ureas in preparative chemistry, see N. Volz and J. Clayden, Angew. Chem., Int. Ed., 2011, 50, 12148 CrossRef CAS PubMed.
- For 4-alkylimino- or 4-arylimino-4H-3,1-benzoxazines, see (a) R. Mazurkiewicz, Monatsh. Chem., 1989, 120, 973 CrossRef CAS; (b) F. He and B. B. Snider, J. Org. Chem., 1999, 64, 1397 CrossRef CAS; (c) B. B. Snider and H. Zeng, J. Org. Chem., 2003, 68, 545 CrossRef CAS PubMed; (d) B. B. Snider and H. Zeng, Heterocycles, 2003, 61, 173 CrossRef CAS; (e) D. Bonne, M. Dekhane and J. Zhu, Org. Lett., 2005, 7, 5285 CrossRef CAS PubMed; (f) K. Aradi and Z. Novák, Adv. Synth. Catal., 2015, 357, 371 CrossRef CAS.
- For a ZnCl2-catalyzed formation of 4-imino-4H-3,1-benzoxazines, see S. Ma, J. Li, Y. Sun, J. Zhao, X. Zhao, X. Yang, L. Zhang, L. Wang and Z. Zhou, Tetrahedron, 2006, 62, 7999 CrossRef CAS.
- U. Neumann and M. Gütschow, Bioorg. Chem., 1995, 23, 72 CrossRef CAS.
- For the formation of 2-amino-4H-3,1-benzothiazin-4-one 7 (R1, R2 = H) from 2-(3-acylthioureido)benzonitriles upon treatment with concd H2SO4 via hydrolysis of the 4-imino moiety, see ref. 11.
- P. Pazdera, V. Potuček, E. Nováček, I. Kalviňš, P. Trapencieris and O. Pugovics, Chem. Pap., 1991, 45, 527 CAS.
- (a) M. Gütschow, M. Schlenk, J. Gäb, M. Paskaleva, M. W. Alnouri, S. Scolari, J. Iqbal and C. E. Müller, J. Med. Chem., 2012, 55, 3331 CrossRef PubMed; (b) A. Stössel, M. Schlenk, S. Hinz, P. Küppers, J. Heer, M. Gütschow and C. E. Müller, J. Med. Chem., 2013, 56, 4580 CrossRef PubMed; (c) Z. Fu, J. He, A. Tong, Y. Xie and Y. Wei, Synthesis, 2013, 45, 2843 CrossRef CAS.
- S. Blättermann, L. Peters, P. A. Ottersbach, A. Bock, V. Konya, C. D. Weaver, A. Gonzalez, R. Schröder, R. Tyagi, P. Luschnig, J. Gäb, S. Hennen, T. Ulven, L. Pardo, K. Mohr, M. Gütschow, A. Heinemann and E. Kostenis, Nat. Chem. Biol., 2012, 8, 631 CrossRef PubMed.
- For the outstanding antimycobacterial properties of nitro derivatives of isomeric 2-amino-4H-1,3-benzothiazin-4-ones, see (a) V. Makarov, G. Manina, K. Mikusova, U. Möllmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. de Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801 CrossRef CAS PubMed; (b) R. Tiwari, G. C. Moraski, V. Krchňák, P. A. Miller, M. Colon-Martinez, E. Herrero, A. G. Oliver and M. J. Miller, J. Am. Chem. Soc., 2013, 135, 3539 CrossRef CAS PubMed.
- For representative reports on 4H-3,1-benzothiazines not belonging to the general structure 7, see, for example (a) A. Hari and B. L. Miller, Org. Lett., 2000, 2, 3667 CrossRef CAS PubMed; (b) W. M. Fathalla and P. Pazdera, Molecules, 2002, 7, 96 CrossRef CAS; (c) P. Langer and U. Albrecht, Synlett, 2003, 10, 1503 CrossRef; (d) F. R. Alexandre, A. Berecibar, R. Wrigglesworth, L. Perreux, J. Guillon, J. M. Léger, V. Thiéry and T. Besson, Tetrahedron, 2005, 61, 8288 CrossRef CAS; (e) Q. Ding and J. Wu, J. Comb. Chem., 2008, 10, 541 CrossRef CAS PubMed; (f) F. Bendrath and P. Langer, Curr. Org. Chem., 2009, 13, 955 CrossRef CAS; (g) C. Gimbert and A. Vallribera, Org. Lett., 2009, 11, 269 CrossRef CAS PubMed; (h) H. Sashida, M. Kaname and M. Minoura, Tetrahedron, 2013, 69, 6478 CrossRef CAS; (i) K. Ezaki, M. Tanmatsu and K. Kobayashi, Heterocycles, 2013, 87, 1311 CrossRef CAS; (j) K. Kobayashi, K. Yamane, I. Nozawa and K. Ezaki, Helv. Chim. Acta, 2014, 97, 315 CrossRef CAS.
- For the preparation of 2-amino-4H-3,1-benzoxazin-4-ones, see (a) E. P. Papadopoulos, J. Heterocycl. Chem., 1984, 21, 1411 CrossRef CAS; (b) J. Bergman, S. Bergman and T. Brimert, Tetrahedron, 1999, 55, 10447 CrossRef CAS; (c) G. M. Coppola, J. Heterocycl. Chem., 2000, 37, 1369 CrossRef CAS; (d) C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830 CrossRef CAS PubMed; (e) A. V. Lygin and A. de Meijere, J. Org. Chem., 2009, 74, 4554 CrossRef CAS PubMed; (f) T. Vlaar, R. V. Orru, B. U. Maes and E. Ruijter, J. Org. Chem., 2013, 78, 10469 CrossRef CAS PubMed; (g) X. F. Wu, M. Sharif, K. Shoaib, H. Neumann, A. Pews-Davtyan, P. Langer and M. Beller, Chem.–Eur. J., 2013, 19, 6230 CrossRef CAS PubMed.
- A. Krantz, R. W. Spencer, T. F. Tam, T. J. Liak, L. J. Copp, E. M. Thomas and S. P. Rafferty, J. Med. Chem., 1990, 33, 464 CrossRef CAS PubMed.
- M. Gütschow and U. Neumann, Bioorg. Med. Chem., 1997, 5, 1935 CrossRef.
- (a) A. Krantz, R. W. Spencer, T. F. Tam, E. Thomas and L. J. Copp, J. Med. Chem., 1987, 30, 489 CrossRef CAS PubMed; (b) N. A. Abood, L. A. Schretzman, D. L. Flynn, K. A. Houseman, A. J. Wittwer, V. M. Dilworth, P. J. Hippenmeyer and B. C. Holwerda, Bioorg. Med. Chem. Lett., 1997, 7, 2105 CrossRef CAS; (c) S. J. Hays, B. W. Caprathe, J. L. Gilmore, N. Amin, M. R. Emmerling, W. Michael, R. Nadimpalli, R. Nath, K. J. Raser, D. Stafford, D. Watson, K. Wang and J. C. Jaen, J. Med. Chem., 1998, 41, 1060 CrossRef CAS PubMed; (d) U. Neumann, N. M. Schechter and M. Gütschow, Bioorg. Med. Chem., 2001, 9, 947 CrossRef CAS PubMed; (e) Z. Y. Ge, Q. M. Xu, X. D. Fei, T. Tang, Y. M. Zhu and S. J. Ji, J. Org. Chem., 2013, 78, 4524 CrossRef CAS PubMed.
- For the 2-alkoxy-4H-3,1-benzoxazin-4-one derivative cetilistat (ATL-962), a lipase inhibitor approved for use in the long-term management of obesity, see P. Kopelman, A. Bryson, R. Hickling, A. Rissanen, S. Rossner, S. Toubro and P. Valensi, Int. J. Obes., 2007, 31, 494 CrossRef CAS PubMed.
- P. Pazdera, J. Meindl and E. Nováček, Chem. Pap., 1992, 46, 322 CAS.
- K. J. Padiya, S. Gavade, B. Kardile, M. Tiwari, S. Bajare, M. Mane, V. Gaware, S. Varghese, D. Harel and S. Kurhade, Org. Lett., 2012, 14, 2814 CrossRef CAS PubMed.
- M. B. Vovk, Russ. J. Org. Chem., 2007, 43, 312 CrossRef CAS.
- For examples of the carbamate-urea transformation, see (a) B. Thavonekham, Synthesis, 1997, 1189 CrossRef CAS; (b) M. Pietsch and M. Gütschow, J. Med. Chem., 2005, 48, 8270 CrossRef CAS PubMed; (c) J. Clayden, L. Lemiègre, M. Pickworth and L. Jones, Org. Biomol. Chem., 2008, 6, 2908 RSC; (d) D. Ke, C. Zhan, X. Li, A. D. Li and J. Yao, Tetrahedron, 2009, 65, 8269 CrossRef CAS.
- The X-ray crystallographic data collection of 12a was performed on a Bruker X8 KappaApex-II diffractometer (CCD) at 100(2)K. The diffractometers were equipped with a low-temperature device (Kryoflex I, Bruker AXS) and used graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Intensities were measured by fine-slicing φ- and ω-scans and corrected for background, polarization and Lorentz effects. Semi-empirical absorption corrections were applied for all data sets by using Bruker's SADABS program. The structures were solved by direct methods and refined anisotropically by the least-squares procedure implemented in the ShelX program system. The hydrogen atoms were included isotropically using the riding model on the bound carbon atoms. The structure was deposited with the Cambridge Crystallographic Data Centre (CCDC-1432796).
- The tautomerizable 2-acetylamino-4-imino-4H-3,1-benzothiazine was reported to undergo acetylation with acetic anhydride not at the imino nitrogen, but at 1-position to 1-acetyl-2-acetylimino-4-imino-4H-3,1-benzothiazine, see ref. 11.
- C. Schickaneder, F. W. Heinemann and R. Alsfasser, Eur. J. Inorg. Chem., 2006, 2357 CrossRef CAS.
- S. V. Downing, E. Aguilar and A. I. Meyers, J. Org. Chem., 1999, 64, 826 CrossRef CAS PubMed.
- M. Gütschow, J. Org. Chem., 1999, 64, 5109 CrossRef.
- (a) R. Leardini, D. Nanni, P. Pareschi, A. Tundo and G. Zanardi, J. Org. Chem., 1997, 62, 8394 CrossRef CAS PubMed; (b) M. Gütschow and U. Neumann, J. Med. Chem., 1998, 41, 1729 CrossRef PubMed.
- M. Gütschow, M. Pietsch, A. Themann, J. Fahrig and B. Schulze, J. Enzyme Inhib. Med. Chem., 2005, 20, 341 CrossRef PubMed.
- M. T. Sisay, S. Hautmann, C. Mehner, G. M. König, J. Bajorath and M. Gütschow, ChemMedChem, 2009, 4, 1425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystal structure (CIF), NMR experiments, calculations of molecular planarity and plane deviations, demonstration of color change in the course of the formation of 9, HRMS-ESI of the product of the treatment of 2c with H218O, enzyme inhibition data, 1H NMR and 13C NMR spectra. CCDC 1432796. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00196c |
| This journal is © The Royal Society of Chemistry 2016 |