
One synthesis: two redox states. Temperature-oriented crystallization of a charge transfer {Fe2Co2} square complex in a {Fe IILSCo IIILS}2 diamagnetic or {Fe IIILSCo IIHS}2 paramagnetic state†
Abstract
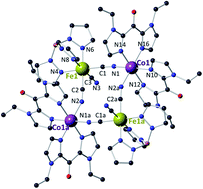
The reaction of [FeIII(Tp)(CN)3]− with [CoII(vbik)2(S)2]2+ leads selectively to the crystallization of cyanide-bridged diamagnetic {FeIILSCoIIILS}2 squares at 5 °C or paramagnetic {FeIIILSCoIIHS}2 ones at 35 °C. The diamagnetic crystal phase can be converted to a paramagnetic one upon heating and it shows photomagnetic effects at low temperature.
 Please wait while we load your content...
Please wait while we load your content...

