
Jacalin-capped silver nanoparticles minimize the dosage use of the anticancer drug, shikonin derivatives, against human chronic myeloid leukemia†
Khan Behlol Ayaz Ahmeda,
Santanu Kar Mahapatrab,
Mamilla R. Charan Rajab,
Shankar Subramaniama,
Megarajan Sengana,
Narendran Rajendrana,
Sandeep Kumar Dasc,
Kuntal Haldard,
Somenath Royc,
Aravind Sivasubramanian *a and
Veerappan Anbazhagan*a
*a and
Veerappan Anbazhagan*a
aDepartment of Chemistry, School of Chemical and Biotechnology, SASTRA University, Thanjavur – 613 401, Tamil Nadu, India. E-mail: anbazhagan@scbt.sastra.edu; anbugv@gmail.com
bMedicinal Chemistry and Immunology Laboratory, Department of Biotechnology, School of Chemical and Biotechnology, SASTRA University, Thanjavur-613 401, Tamil Nadu, India
cImmunology and Microbiology Laboratory, Department of Human Physiology with Community Health, Vidyasagar University, Midnapore-721 102, West Bengal, India
dDivision of Molecular Medicine, Bose Institute, Kolkata 700 054, India
First published on 4th February 2016
Abstract
Repeated consumption of a chemotherapeutic drug in high doses often leads to drug resistance. The objective of this study was to develop a facile method to enhance the anticancer activity of the phytomolecules, acetylshikonin (AS) and beta,beta-dimethylacrylshikonin (BDS). Herein, we demonstrated that jacalin-capped silver nanoparticles (JAgNPs) loaded with AS/BDS induce maximum cytotoxicity effects on human chronic myeloid leukemia (CML), K562 at low concentration (100 nM), whereas for a similar effect about 500 nM of pure AS/BDS was required. Fluorescence microscopy data revealed the internalization of JAgNPs-AS/BDS complex into K562 cells. The intracellular localization of the drug caused the production of excess reactive oxygen species (ROS), elevation in the secretion of tumor necrosis factor (TNF-α), suppression in the production of interleukin-10 (IL-10), losses of mitochondrial membrane potential, DNA damage and apoptosis. The effective role of ROS and TNF-α in JAgNPs-AS/BDS mediated cell death was proven by pretreatment of cells with N-acetyl cysteine, a ROS scavenger, and pentoxifylline, a potent TNF-α blocker. The mode of K562 cell death was confirmed by annexin-FITC/PI staining followed by flow-cytometric analysis. Further, we disclosed the contribution of different caspase activation pathways in TNF-α mediated cell death. Taken together, our study elucidated that the judicious use of AgNPs might improve the therapeutic efficacy of AS/BDS against CML at lower doses.
1. Introduction
During the last decade, medical science has witnessed considerable advancement in understanding and developing a new therapeutic modality for treating various cancers. Despite these developments, the world-wide human death toll increases every year due to low effectiveness of currently available cancer treatments.1,2 Major issues in cancer treatment are the side-effects caused by non-specific drug delivery, which results in non-specific damage to even visceral organs.3 An additional drawback is that half of all chemotherapy patients develop drug resistance due to repeated consumption of a drug in high dosages.4–6 Hence, there is an urgent need to identify safer compounds and optimize drug efficacy at a lower effective dose and also improve selective uptake into cancerous cells.Plant extracts are widely used in traditional Chinese and Indian medicine.7–10 Incidentally, many of the currently used chemotherapeutic agents are derived either from plants or are semi-synthetic in nature.11–14 Shikonin, a natural naphthoquinone pigment, was used in traditional Chinese medicine for the treatment of different inflammatory diseases.15 The anticancer activity of shikonin was first reported against murine stroma 180.16 Guo et al. showed that a shikonin-containing mixture was safe and effective in the treatment of 19 patients with late-stage lung cancer.17 Shikonin and its analogs exhibit broad-spectrum activity and could circumvent drug transporter and apoptotic defect mediated cancer drug resistance.18 However, the mechanism of action of shikonin remains enigmatic. In this study, we isolated two naphthoquinones namely acetylshikonin (AS) and β,β-dimethylacrylshikonin (BDS) from Arnebia nobilis and studied them against K562 cells. In the Indian system of medicine, Arnebia nobilis is known as “Rathanjot” and is useful for treating the diseases of eyes, bronchitis, abdominal pains, itch, etc.19
Metallic medicines are widely used in the Indian system of medicine (ayurveda) to treat various ailments,20 and also in modern medicine metallic based complexes such as cisplatin are used for the treatment of cancer.21 Inspired by traditional Indian medicine, for the first time, we attempted to enhance the anticancer activity of shikonin at a lower dosage by loading it into lectin-capped AgNPs. The choice of lectin was made because of the highly specific lectin/sugar epitope interaction; moreover, many lectins are of non-immune origin.22 Maackia amurensis lectin has been used to target terminal α 2,3-sialylated O-linked glycans of melanoma cells.23 Agaricus bisporus lectin has specificity for GlcNAC-exposed N-linked glycans and reversibly inhibits the proliferation of MCF-7 breast cancer cells, Caco-2 colon cancer cells, and HT-29 colon cancer cells.24 Morniga G lectin conjugated with porphyrin photosensitizer was used to target the T-antigen and the Tn antigen expressed on Jurkat leukaemia cells.25 Peanut agglutinin loaded with fluorescence nanospheres was used to detect orthotopically implanted colorectal tumors expressing the T-antigen.26
Silver nanoparticles (AgNPs) are well-known for their cytotoxic properties27 and were expected to act synergistically in the presence of chemotherapeutic agents. Herein, we synthesized jacalin-capped AgNPs to boost anticancer activity of the shikonin derivatives, acetylshikonin (AS) and beta,beta-dimethylacrylshikonin (BDS). Jacalin, a lectin isolated from the seed of Artrocarpus integrifolia, is highly specific to T-antigen disaccharide (2-acetamido, 2-deoxy, 3-O-β-D-galactopyranosyl-α-D-galactopyranoside), which is over expressed in 85% of carcinomas.28,29 Marangoni et al. showed that jacalin-gold nanoparticles conjugate exhibit high affinity for leukemic K562 cells.30 Obaid et al. directly coupled jacalin to PEG phthalocyanine gold nanoparticles to target T-antigen expressed on HT-29 colon adenocarcinoma cells.31 Other than specific sugar, jacalin is known for binding other molecules such as porphyrin, phycocyanin, ruthenium complexes, AgNPs, etc.32–36 Hence, jacalin can serve both as hydrophobic and hydrophilic drug carrier and can be used for drug targeting.
In this paper, we report the interaction between jacalin and AS/BDS using fluorescence spectroscopy. Interaction studies in the presence of galactose and agglutination inhibition assay revealed that the T-antigen binding site of jacalin was preserved even after binding to AS and BDS. Thus, we prepared AS and BDS conjugates with jacalin-capped AgNPs and tested against human chronic myeloid leukemia (CML), K562. To establish the anticancer mechanism, we checked ROS generation and cytokines release assay, which were further confirmed by specific inhibitor molecules, and finally shikonin-derivatives-induced apoptosis of K562 cells was confirmed by annexin V-FITC/PI staining followed by FACS analysis.
2. Experimental
2.1. Binding of AS/BDS to jacalin
Jacalin was purified by affinity chromatography on cross-linked guar gum as described previously.32,37 Acetylshikonin and β,β-dimethylacrylshikonin were isolated from Arnebia nobilis with established protocols.38,39The interaction studies were performed in a Jasco-FP8200 spectrofluorometer with a spectral slit width set to 2.5 nm for both excitation and emission monochromators. The intrinsic fluorescence spectra of jacalin were recorded from 300–400 nm at an excitation wavelength of 280 nm. AS/BDS were dissolved in DMSO and used as is. A fixed volume of jacalin solution (3.0 mL, 5 μM) was titrated by adding small aliquots of the AS/BDS from a concentrated stock solution (0.4 μM) and the fluorescence intensity was recorded after an equilibration period of 2 min. To determine if binding of AS/BDS interferes with the natural saccharide binding characteristic of jacalin, we performed NPs interaction studies by pre-incubating jacalin with a high concentration (100 mM) of galactose. All binding experiments were performed in PBS (phosphate buffered saline) buffer. All titrations were repeated at least three times to arrive at average values.
2.2. Preparation of jacalin-capped AgNPs and AS/BS loaded jacalin-capped AgNPs
In a typical procedure to synthesize AgNPs, 1 mM silver nitrate (AgNO3) and 5 μM of jacalin was dissolved in PBS buffer and stirred for 2 min. Then, 1 mm of NaBH4 was added into the above solution. The solution was initially colorless and it turned to a yellow color after 10 min, indicating the formation of AgNPs. AS/BS loaded jacalin-capped AgNPs were prepared by treating the 1 mg mL−1 AS/BDS-5 μM jacalin-1 mM AgNO3 complex with 1 mM NaBH4. The activity of the lectin and lectin–drug complex was checked by hemagglutination and hemagglutination inhibition assays as described in ref. 40 and 41.The optical absorbance of the synthesized jacalin-capped AgNPs and AS/BS loaded jacalin-capped AgNPs was monitored by a UV-vis spectrophotometer (Thermo Scientific Evolution 201) between wavelengths of 300 to 800 nm at a resolution of 1 nm. Size and morphology were examined using transmission electron microscopy (JEOL-JEM 1011, Japan) operating at an accelerated voltage of 200 kV.
2.3. Cytotoxicity
The MTT (3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide) assay was performed to estimate viability of K562 cells in the presence of AS, BDS, jacalin-bound AS (JAS), jacalin-bound BDS (JBDS), jacalin-capped AgNPs-bound AS (AgJAS) and jacalin-capped AgNPs-bound BDS (AgJBDS). Briefly, 2 × 104 K562 cells per well, cultured in RPMI 1640 medium supplemented with 10% FBS and 1× penicillin–streptomycin, and 50 μg mL−1 gentamycin, were placed in a 5% CO2 incubator (NBS-Eppendorf, Galaxy 170S) at 37 °C. The cells were treated with different concentrations of shikonin derivatives (10, 50, 100, 250, 500, 750, and 1000 nM) for 24 h at 37 °C. After that, 10 μL MTT solution (5 mg mL−1) was added per well for 4 h followed by solubilizing in a HCl–isopropanol solution. The absorbance of MTT formazan product was measured using a microplate reader (Biotek, Synergy H1, India) at 570 nm. Cell viability was calculated as a relative percentage to untreated cells.42 Similarly, cell viability was estimated at a different time point of shikonin derivatives treatment to K562 cells by MTT assay. In a separate set of experiments, human peripheral blood lymphocytes (PBL) were isolated from heparinized blood followed by treated with shikonin derivatives or jacalin-capped AgNPs for 24 h and cell cytotoxicity was measured by the MTT assay. All the data presented in this paper were collected from similar experiments repeated at least three times and expressed as mean ± standard deviation.2.4. Intracellular measurement of reactive oxygen species (ROS)
The production of intracellular ROS was measured using 2,7-dichlorofluorescein diacetate (DCFH2-DA).43 Briefly, 10 mM DCFH2-DA stock solution (in methanol) was diluted in culture medium without serum or other additives to yield a 100 μM working solution. At the end of exposure with shikonin derivatives, cells were washed twice with PBS. Then, cells were incubated in a 1.5 mL working solution of DCFH2-DA at 37 °C for 30 min. Cells were lysed in alkaline solution and centrifuged at 3000 rpm. One milliliter of supernatant was transferred to a cuvette, and fluorescence was measured at 520 nm with a fluorescence spectrophotometer (Jasco-FP8200, Japan) using 485 nm excitation. As a positive control, some of those cells were incubated with H2O2 (100 mM) for 30 min prior to the analysis. The values were expressed as percent fluorescence intensity relative to control wells. Fluorescence micrographs were also taken by phase contrast microscopy (NIKON ECLIPSE LV100POL, Japan). To determine the crucial cues of ROS generation in shikonin derivatives-induced cell death, K562 cells (2 × 104) were seeded in a 96-well plate with 0.2 mL per well. A stock solution of N-acetyl-L-cysteine (NAC) was made with sterile water and added to cells at 10 mM for 1 h.42 NAC pre-treated or untreated K562 cells were cultured with 100 nM shikonin derivatives for 24 h and cell viability was determined by the MTT method.422.5. Lactic dehydrogenase (LDH) releases assay
LDH release was estimated using a spectrophotometric LDH assay kit (Reckon Diagnostics Pvt. Ltd, India) to evaluate cell membrane integrity. The shikonin derivatives (100 nM) induced LDH released by the K562 cells (2 × 104 cells per well) found in the culture supernatants was determined by spectrophotometric analysis according to the manufacturer's instructions. To study the involvement of necrosis in the LDH release, we pre-treated K562 cells with 60 μM necrostatin-1 (inhibitor of necrosis) for 1 h prior to the treatment of shikonin derivatives (100 nM), cultured them for 24 h, and then followed the cell viability by MTT assay.442.6. Apoptotic morphological changes by AO/Et–Br and DAPI staining
Two DNA-binding dyes (AO and Et–Br) were used with the morphological apoptotic cells. After treatment with 100 nM shikonin derivatives for 24 h, K562 cells were collected, washed with cold PBS, and then stained with a mixture of AO (100 μg mL−1) and Et–Br (100 μg mL−1) at room temperature for 5 min in the dark. After proper washing with PBS, the stained cells were observed using a fluorescence microscope (NIKON ECLIPSE LV100POL, Japan).Untreated and shikonin derivatives treated K562 cells were collected and fixed with 2.5% glutaraldehyde for 15 min, followed by permeabilization with 0.1% Triton X-100 and stained with 1 μg mL−1 DAPI for 5 min at 37 °C. The cells were then washed with PBS and examined by fluorescence microscopy (NIKON ECLIPSE LV100POL).
2.7. Assessment of apoptotic cell population by FITC-annexin V/PI double-staining assay
Cells (2 × 106) were seeded in 35 mM cell culture plates and treated with shikonin derivatives (100 nM) for 24 h. Then the K562 cells were collected, washed with PBS, and re-suspended in PBS. Apoptotic cell death was identified by double staining with recombinant FITC-conjugated annexin V and PI, using the annexin V-FITC apoptosis detection kit (BD Bioscience, India) according to the manufacturer's instructions. Flow cytometric analysis was performed immediately after dual staining. Data acquisition and analysis were performed in a Becton-Dickinson FACS verse flow cytometer using CellQuest software.2.8. Cytokine analysis
Shikonin derivatives induced cytokines (TNF-α and IL-10) productions were determined from cell free supernatants of culture media with an enzyme-linked immunosorbent assay (ELISA) (e-Bioscience, India) according to the manufacturer's instructions.K562 cells were pre-treated with 2 mM pentoxifylline (a potent TNF-α inhibitor) and 30 μM SP600125 (JNK inhibitor) and then treated with shikonin derivatives (100 nM) for 24 h. After the treatment schedule, the cells were washed with culture medium and cell viability was estimated with a MTT assay.42
2.9. Drug uptake assay
AgJAS and AgJBDS uptakes by K562 cells were studied by fluorescence microscopy imaging (NIKON ECLIPSE LV100POL). Briefly, 50 μL rhodamine B (2 mg mL−1) and AgJAS/AgJBDS, mixed and stirred for 24 h at 37 °C, and unbound rhodamine B were removed by repeated washing. K562 (2 × 104) cells were plated in 6-well chambered glass slides in presence of freshly prepared Rh-B labelled AgJAS/AgJBDS (100 nm) and were incubated for 4 h at 37 °C in a CO2 incubator. After that, the cells were washed 2 times with PBS and immediately observed under the fluorescence microscope for uptake assessment. Images were acquired at 50× optical zoom and analysis was done using ImageJ software v.r. 1.43.3. Results and discussion
3.1. Interaction of jacalin with AS and BDS
The primary aim of this study is to enhance the anticancer activity of shikonin derivatives AS and BDS by loading them into jacalin-capped AgNPs. For this study, the naphthoquinones AS and BDS were isolated from A. nobilis and characterized comprehensively (Fig. S1†). Proton NMR data was in complete concurrence with the reported values.38,39 Optical rotation studies unequivocally proved that the naphthoquinones isolated were shikonins and not alkanins.45The interaction between a drug and the carrier is very important for therapeutic efficacy. Therefore, first we investigated the interaction between jacalin and shikonin derivatives using the intrinsic fluorescence of jacalin and hemagglutination assay. Fluorescence spectra of jacalin in the absence and presence of different concentrations of AS or BDS are shown in Fig. 1. From these spectra, it is clear that the addition of drug decreases the fluorescence of jacalin without a shift in the fluorescence emission maximum. It suggests that AS and BDS bind to jacalin without affecting their secondary structures. This was further supported by the far-UV circular dichroism, which showed no substantial changes in the protein secondary structure upon drug binding [data not shown]. A binding curve depicting progress of the titration of the drug with jacalin is shown in Fig. S2.† Increasing the drug concentration leads to an increase in the change in fluorescence intensity (ΔF); however, the magnitude of the change decreases with increasing drug concentration and this displays saturation behavior at high concentrations of the drug. From this binding data the association constant, Ka was determined by plotting log[(F0 − Fc/Fc − F∞)] against log[drug] (inset of Fig. S2†). The Ka value for the jacalin-AS (JAS) and jacalin-BDS (JBDS) interaction is determined as 7.94 × 104 M−1 and 6.13 × 104 M−1, respectively.
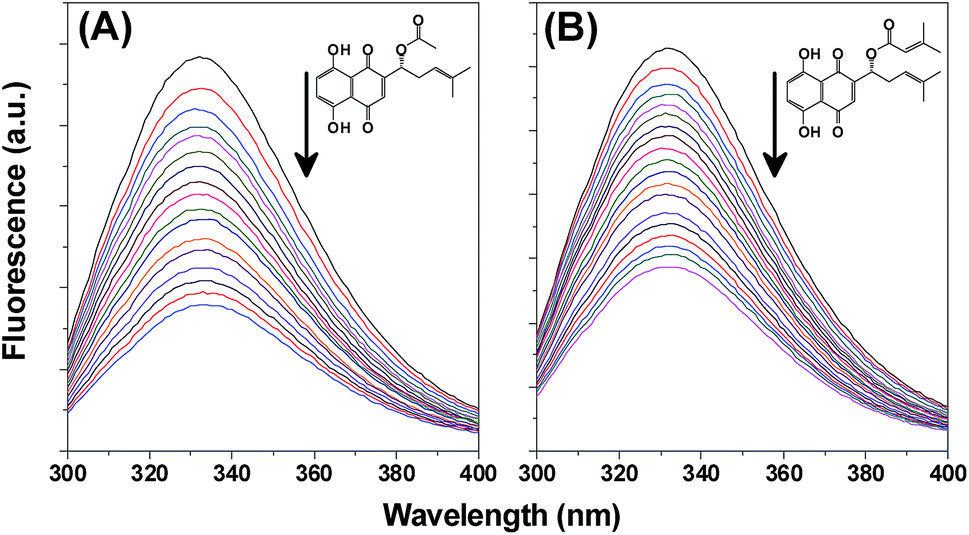 | ||
| Fig. 1 Jacalin fluorescence spectra monitored after addition of increasing concentrations of (A) acetyl shikonin and (B) beta,beta-dimethylacrylshikonin. The upper spectrum in each panel corresponds to free jacalin and the remaining spectra with decreasing fluorescence emission were obtained in the presence of increasing concentrations of AS/BDS. Insets correspond to structures of (A) AS and (B) BDS. | ||
The sugar binding site of jacalin is very important for T-cell recognition. Therefore, we investigated the interaction of AS and BDS in the presence of 100 mM galactose. Following the above described method, the Ka value for the jacalin and AS and BDS interaction in the presence of galactose was determined as 4.36 × 104 M−1 and 4.59 × 104 M−1, respectively. The slight variation in the binding constant in the presence of galactose can be accounted for by the conformational changes in jacalin upon sugar binding.46 Hemagglutination experiments have been widely used to validate the functional nature of a lectin.40,41 Agglutination inhibition assay indicated no loss of erythrocyte-agglutination activity by jacalin, JAS, and JBDS complex [Fig. 2A, lane ii, iv, and v]. However, the addition of 100 mM galactose to JAS and JBDS complex inhibits agglutination suggesting that the binding sites on jacalin for the AS/BDS and sugar are distinctly different [Fig. S3†]. These results also indicate that jacalin remains in the active functional state while interacting with AS and BDS. The binding constant obtained here for the jacalin-AS/BDS complexes are in the range of 104 M−1, which are comparable to those observed generally for lectin–carbohydrate and lectin–non-carbohydrate complexes.32–36 The moderately high binding constant observed here indicates that the maximum percentage of the shikonin derivatives is expected to be in the lectin-bound state. Therefore, these conjugates are expected to deliver larger amounts of drug specifically to the target tissues.
 | ||
| Fig. 2 (A) Hemagglutination assays with human erythrocytes. Lane (i) DMSO, (ii) jacalin, (iii) jacalin–galactose complex, (iv) JAS, (v) JBDS, (vi) JAgNPs, (vii) AgJAS, (viii) AgJBDS, (ix) AgJAS–galactose complex, and (x) AgJBDS–galactose complex. About 100 μL of 4% human erythrocytes are used. Jacalain agglutinates erythrocytes whereas the presence of 100 mM galactose inhibits the agglutination. The agglutination was only inhibited by the specific sugar galactose and not by AS/BDS (lane iii). Experiments were performed in triplicate. (B) UV-visible spectra of jacalin-capped AgNPs (solid line), AS loaded jacalin-AgNPs (dashed line) and BDS loaded jacalin-AgNPs (dotted line). The stock concentration of AS and BDS was 1 mM. Inset corresponds to the photograph of (a) jacalin-AgNPs, (b) AgJAS, and (c) AgJBDS. (C) TEM images of JAgNPs, (D) AgJAS, and (E) AgJBDS. Inset corresponds to the SAED pattern. | ||
3.2. Preparation of jacalin-AgNPs and jacalin-AgNPs–AS/BDS complexes
Previous studies showed that the binding sites on jacalin for the AgNPs and sugar are distinctly different. Therefore, we directly synthesized jacalin-capped AgNPs (JAgNPs) as described in material and methods and loaded the shikonin derivatives. JAgNPs had a dark yellow color and showed the characteristic surface plasmon resonance (SPR) maximum at 440 nm. Addition of AS and BDS to JAgNPs shifted the SPR maximum to 425 and 427 nm, respectively (Fig. 2B). The observed difference in the SPR maximum can be accounted for by change in the polydispersity of the JAgNPs suspension. It is well known that the position and shape of the AgNPs SPR peak depend on particle size, shape, and dielectric constant of the surrounding medium.47 TEM images of JAgNPs, AS-loaded JAgNPs (AgJAS), and BDS-loaded JAgNPs (AgJBDS) are shown in Fig. 2C–E. The size of the AgNPs prepared with jacalin, AgJAS, and AgJBDS are in the range of 6–18 nm, 3–25 nm, and 2–24 nm, respectively. A background of protein layer was observed in all the TEM images, accounting for stabilization of the AgNPs. Selected area electron diffraction (SAED) shows a ring pattern, supporting the formation of crystalline AgNPs (inset of Fig. 2).Since the sugar binding site of jacalin is very important for T-cell recognition, we verified the functional nature of the AgJAS and AgJBDS complex by hemagglutination assay (Fig. 2A). Both AgJAS and AgJBDS complexes showed hemagglutination [Fig. 2A, lane vii and viii], which was inhibited by the addition of 100 mM galactose [Fig. 2A, lane ix and x]. These results suggest that the formation of AgJAS and AgJBDS complexes has no interference with the jacalin sugar binding site. Our data suggest that the sugar binding site is free; therefore, we expected that AgJAS and AgJBDS can recognize T-cells without any trouble. However, it may be argued that the cell agglutination nature of jacalin can lead to massive and instantaneous clotting of blood, when a shikonin–jacalin mixture is injected intravenously, resulting in severe, inadvertent damage to the subject. Indeed, ConA-daunomycin conjugate has been proven to be effective when injected intraperitoneally to tumor-bearing mice, thus circumventing the negative effects associated with agglutination.48 Moreover, the presence of AgNPs in our formulation may facilitate entry by exploiting the leakage vasculature of the tumorous tissues. Therefore, we envisage that a judicious use of jacalin-AgNPs–shikonin complexes will be useful in increasing the drug efficacy in chemotherapeutics.
3.3. Cytotoxicity of shikonin derivatives
MTT assay was performed to assess the cytotoxicity of shikonin derivatives toward CML, K562. All the tested shikonin derivatives (AS, BDS, JAS, JBDS, AgJAS, and AgJBDS) induced cell death in a concentration and time-dependent manner. Strikingly, AS, BDS, JAS, and JBDS induced significant cell death from 50 nM and reached a plateau at 500 nM, whereas AgJAS and AgJBDS induced significant cell death even at lower concentration (10 nM) and reached the plateau at 100 nm. The control experiments performed with jacalin-capped AgNPs showed negligible cytotoxicity, indicating that significant cytotoxicity was induced by AS/BDS (Fig. S4†). However, the observed enhanced cytotoxicity of the drug loaded JAgNPs may be due to transport of more drugs into the K562 cells by the JAgNPs exploiting the leaky vasculature of the cancer cell.It is noteworthy that 500 nM of AS and BDS induced maximum cell death (89% and 73% respectively) after 24 h, while AgJAS and AgJBDS obtained the similar activity (88% and 77%, respectively) at 100 nM concentration after 24 h (Fig. 3A). The IC50 value of AS, BDS, JAS, JBDS, AgJAS, and AgJBDS against K562 cells were found to be 164.7 nM, 172.4 nM, 142.7 nM, 155.54 nM, 51.33 nM and 61.76 nM respectively, while IC50 of DOX is 4.98 μM (2.9 μg mL−1).42
 | ||
| Fig. 3 In vitro shikonin derivatives induced concentrations dependent and time dependent cell death of K562 cells. (A) K562 cells were treated with 0–1000 nM shikonin derivatives for 24 h (B) K562 cells were treated with 100 nM shikonin derivatives for indicated time period. % of cell death was measured by MTT assay. The experiments were repeated three times, yielding similar results, and data are expressed as means ± SEM. *, p < 0.05; **, p < 0.01 significant difference compared with respective untreated control. | ||
Time-dependent cytotoxicity studies performed using 100 nm of shikonin derivatives revealed that AgJAS and AgJBDS induced considerably higher percentage of cell death as compared to AS, BDS, JAS, and JBDS (Fig. 3B). These results clearly showed that AgNPs plays the crucial role in enhancing the cytotoxicity of shikonin derivatives at very low concentration. The control experiments with human peripheral blood lymphocytes (PBL) could not identify significant cell death at 100 nM of shikonin derivatives (Fig. S5A†). To test the compatibility of the shikonin derivatives with human erythrocytes, we investigated the hemolytic activity of the shikonin derivatives. Hemolytic activity estimated through free hemoglobin showed that all the drugs display negligible hemolysis, which is significantly (p < 0.001) less compared with positive control (Fig. S5B†). These results suggest that the shikonin derivatives are compatible with circulatory lymphocytes and erythrocytes. Based on these data, the physiological safer concentration (100 nm) of shikonin derivatives was selected and used for further studies against CML.
3.4. Generation of reactive oxygen species after exposure to shikonin derivatives
Reactive oxygen species (ROS) are formed as a natural by-product of normal metabolism and play crucial roles in cell signaling and homeostasis. However, an increase in the intracellular ROS may result in significant damage to cell morphology, which ultimately leads to cell death.43,49 DCFH2-DA is widely used as an oxidant-sensitizer, is non-fluorescent, and switches to a highly fluorescent DCF when oxidized by ROS and other peroxides. The addition of DCFH2-DA to the drug-treated cells showed strong fluorescence at 520 nm, whereas the untreated cells and cells treated with JAgNPs showed poor fluorescence, indicating generation of ROS in the drug-treated cells. As noted from Fig. 4A, shikonin derivatives could stimulate ROS production in K562 cells similar to the positive control H2O2.42 Compared to the untreated cells, those treated with AgJAS and AgJBDS at 100 nM had a 7-fold increase of ROS generation, whereas AS, BDS, JAgNPs, JAS, and JBDS had only a 3-fold increase in ROS generation (Fig. 4A), suggesting that JAgNPs in combination with AS/BDS promotes higher production of ROS and enhances the cytotoxicity. However, K562 cells treated with a well-known antioxidant agent, N-acetyl-cysteine (NAC), circumvented the shikonin derivatives-induced ROS generation and thereby decreased cell death caused by the shikonin derivatives (Fig. 4B). Further, the AgJAS and AgJBDS induced ROS generations in K562 cells were confirmed by fluorescence microscopy (Fig. 4C and S6†), and that corroborates the ROS generation estimated by the spectrofluorometric method. | ||
| Fig. 4 (A) Shikonin derivatives-induced ROS generation in K562 cells. (B) Effect of NAC (ROS inhibitor) on shikonin derivatives-induced cell cytotoxicity. (C) Fluorescence microscopy images showing the qualitative ROS generation by shikonin derivatives in K562 cells. Fluorescence microscopy images were measured in the presence of DCFH-DA. (i) Untreated cells, (ii) treated with AgJAS, and (iii) treated with AgJBDS. (D) Shikonin derivatives-induced LDH release by K562 cells. (E) Effect of necrostatin-1 (necrosis inhibitor) on shikonin derivatives-induced cell cytotoxicity. The experiments were repeated three times, yielding similar results, and data are expressed as means ± SEM. *, p < 0.05; **, p < 0.01 significant difference compared with respective untreated control. +ve control are the cells treated with H2O2. | ||
The generation of ROS could decrease plasma membrane integrity and release lactic dehydrogenase (LDH), an important cellular marker for cell viability.50 As shown in Fig. 4D, exposure of K562 cells to 100 nM shikonin derivatives to K562 cells for 24 h resulted in significant (p < 0.05) release of LDH as compared with untreated cells. Increase of LDH leakage in the medium illustrated that the exposure of K562 cells to shikonin derivatives could cause rapid cell lysis, and as a result, membranes become leaky and induce cell death.50 Strikingly, AgJAS and AgJBDS showed a higher percentage of LDH release as compared to AS, BDS, JAgNPs, JAS, and JBDS, augmenting the MTT and ROS assay.
It is well known that the generation of excess ROS could cause cell death by two different pathways, either apoptosis or necrosis. Necrostatin I, an inhibitor of necrosis, was used to evaluate the necrosis mediated cell death in the presence of shikonin derivatives. As shown in Fig. 4E, pre-treatment with necrostatin 1 could not inhibit the shikonin derivatives induced cell death, suggesting that the cell death is not mediated by necrosis, but rather by apoptosis. It is noteworthy that AgJAS and AgJBDs showed a lesser percentage of viable K562 cells as compared to AS, BDS, JAgNPs, JAS, and JBDS, indicating enhanced cytotoxicity of AS and BDS was caused by the presence of AgNPs. These results suggest that excess production of ROS by the shikonin derivatives, AgJAS and AgJBDS, could result in apoptosis through oxidative damage, which are considered a major source of spontaneous damage to DNA and membranes.51
Since mitochondria are the major sites of ROS production inside cells, we further investigated whether ROS generation induced by shikonin derivatives were involved with mitochondrial membrane potential (MMP) changes. It is well established that altered mitochondrial function was linked to apoptosis and the early event of decreased MMP is associated with mitochondrial dysfunction.52 MMP in K562 cells was estimated using the fluorescence probe, rhodamine 123. Our results indicated that shikonin derivatives decreased the MMP significantly (p < 0.05) in K562 cells [Fig. S7†], corroborating others.53 Interestingly, AgJAS and AgJBDS have shown significantly less MMP (as good as standard drug doxorubicin) [Fig. S7†]. These results suggested that the AgNPs present in the JAS and JBDS complexes boosted the pro-apoptotic role of our studied drugs in K562 cells.
3.5. Induction of apoptosis by shikonin derivatives
Induction of apoptosis is the most important key event and also most studied for anticancer strategy. In the present study, induction of apoptosis in leukemic cells by shikonin derivatives was monitored by Et–Br/AO and DAPI staining. As shown in Fig. 5A–C and S8A,† AO was taken up by both viable and non-viable cells and emits green fluorescence,49 whereas Et–Br was taken up only by non-viable cells and emits orange-red fluorescence because of the intercalation into DNA.49 From Fig. 5B and C, it is clear that K562 cells treated with AgJAS and AgJBDS showed orange-red fluorescence suggesting the presence of non-viable cells. In addition, DAPI staining showed that K562 cells exposed to AgJAS and AgJBDS produced an increase in nuclear fragmentation and chromatin condensation [Fig. 5D and E], which provides strong evidence for the apoptotic pathway of cell death.42 It is noteworthy that the K562 cells exposed to AS, BDS, JAS, and JBDS showed a greater number of viable cells as compared to the AgJAS and AgJBS, supporting the role of AgNPs in enhancing the apoptotic activity of AS and BDS [Fig. S8†]. | ||
| Fig. 5 Qualitative analysis of nuclear morphology by (A–C) EtBr/AO and (D–F) DAPI staining using fluorescence microscopy. (A) and (D) are control untreated cells, (B) and (E) are cells exposed to AgJAS and (C) and (F) are cells exposed to AgJBDS. Cells were visualized under fluorescence microscopy at excitation/emission wavelengths 490/620 nm and 345/455 nm respectively. | ||
Further, annexin V-FITC/PI dual staining assay was carried out to evaluate the extent and mode of cell death. Untreated cells and treated cells were sequentially stained with annexin V-FITC and PI and then analyzed through flow cytometry.42 Fig. 6 shows that untreated cells and cells treated with JAgNPs had negligible binding to annexin V-FITC and PI, indicating the presence of viable cells. However, AgJAS/AgJBDS treated cells showed significant population (65% and 61%, respectively) of K562 cells by staining with annexin V-FITC and PI, indicating these cells were undergoing late stage apoptosis. It is noteworthy that the drug-treated cells responded negatively to single staining with PI, indicating the K562 cells were undergoing natural senescence, consistent with the necrosis inhibitor assay. Indeed, the dual staining experiments performed with AS, BDS, JAS, and JBDS showed fewer apoptotic cells as compared to the AgJAS/AgJBDS [Fig. S9†]. These results confirmed the pivotal role of AgNPs in inducing apoptosis in K562 cells.
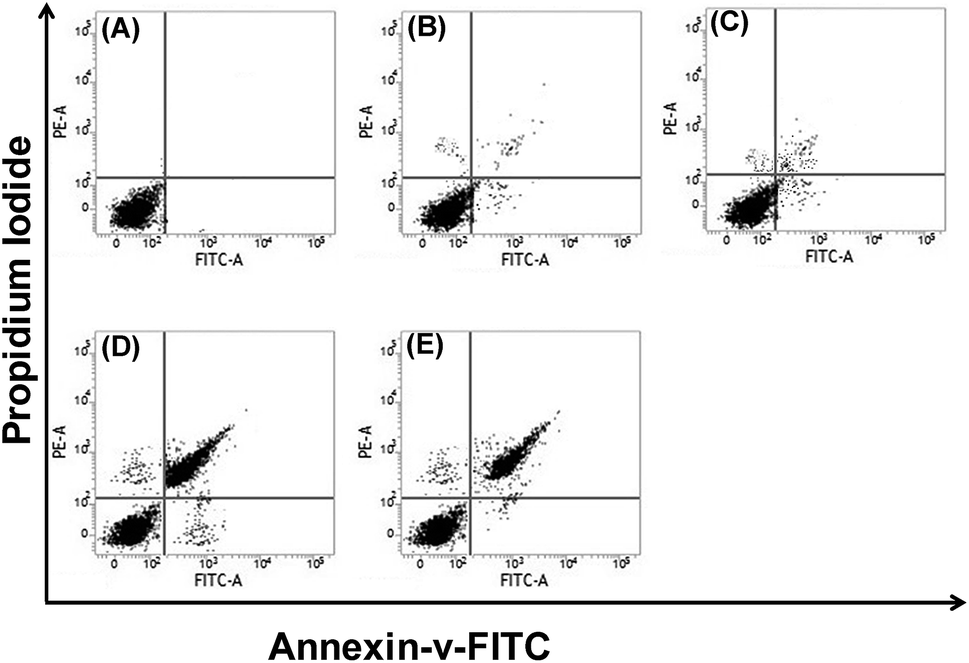 | ||
| Fig. 6 Shikonin derivatives induced apoptosis in K562 cells by annexin-V-FITC/PI dual staining analyzed by FACS. (A) Isotype control, (B) untreated control, (C) JAgNPs, (D) AgJAS, and (E) AgJBDS. | ||
Apoptosis is a tightly regulated process involved with induction of caspase cascade. Fig. 7 shows that K562 cells treated with drug produced significant (p < 0.05) amounts of caspase-9, caspase-8, and caspase-3 activity as compared with untreated K562 cells. Moreover, we observed AgNPs containing formulations of AS and BDS were more efficient inducing the caspase activation in K562 cells. These results suggested the activation of apoptosis by shikonin derivatives was through caspase activation, which is consistent with a previous study.54,55
 | ||
| Fig. 7 Shikonin derivatives-induced caspase activation in K562 cells studied by ELISA. After the treatment with shikonin derivatives (100 nM), the cell lysates were used to quantify the caspase 9, caspase 8, and caspase 3 by ELISA (e-Biosciences, US). The experiments were repeated three times, yielding similar results, and data are expressed as means ± SEM. *, p < 0.05; **, p < 0.01 significant difference compared with respective untreated control. | ||
The inflammatory cytokines, and specifically TNF-α, are known for promoting the caspase 8–caspase 9–caspase 3 dependent apoptotic pathway.56 In normal circumstances, the higher expression of TNF-α is opposed by the higher expression of IL-10, which is crucial for cell survival.57 As inferred from Fig. 8A and B, the AgNPs containing shikonin derivatives significantly increased the TNF-α and decreased the IL-10 release from K562 cells. It was well established that TNF-α is highly responsible for the generation of ROS and activates the downstream apoptosis signals, including NF-κB activation and MAP kinase activation.49 This TNF-α expression could be completely prevented by pre-treatment with the TNF-α inhibitor, pentoxifylline. As a result there was a significant increase in the viable cells (Fig. 8C). Mao et al. reported that shikonin enhanced JNK phosphorylation to induce the apoptosis in CML cells.58 In the current study, lower production of IL-10 observed in the drug treated cells [Fig. 8B] could be associated with JNK phosphorylation. In order to verify that hypothesis, the cells were pre-treated with JNK inhibitor (SP600125), then exposed to shikonin drugs, and the cell viability was followed by MTT assay. SP600125 treated cells significantly (p < 0.05) enhanced the cell viability confirming the involvement of JNK MAP kinase activation, which resulted in the decreased production of IL-10 (Fig. 8D). These results suggested that tested shikonin drugs enhanced the pro-inflammatory cytokines and simultaneously decreased the anti-inflammatory cytokines, which are crucial cues to induce apoptosis signaling in K562 cells.
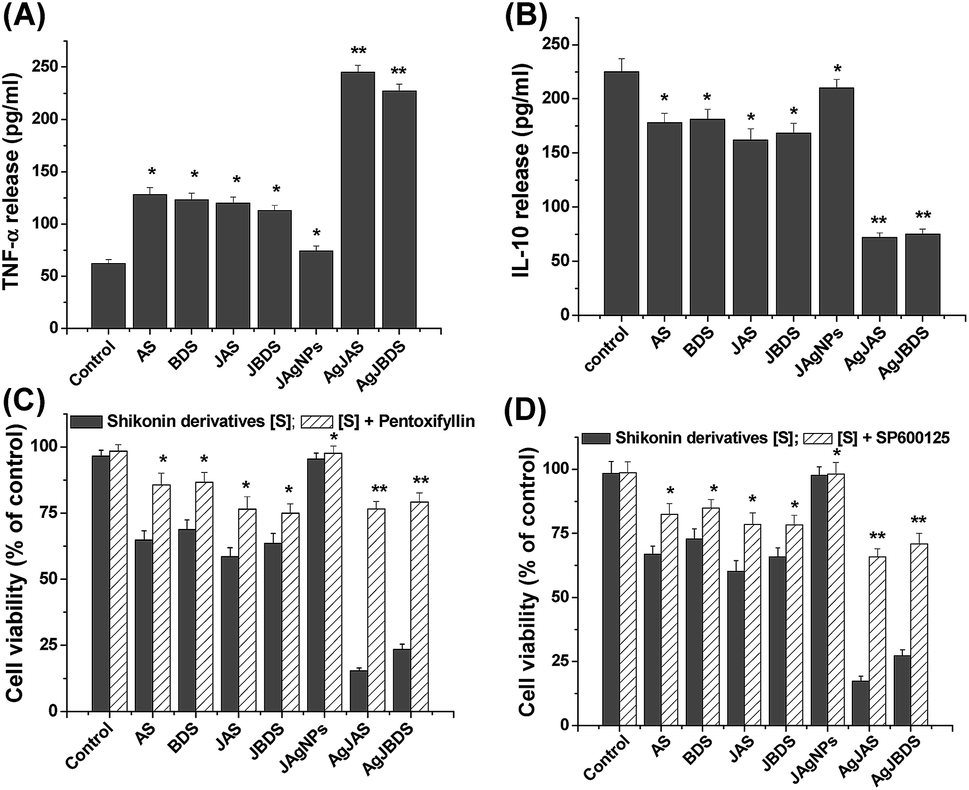 | ||
| Fig. 8 Shikonin derivatives induced alteration of (A) TNF-α and (B) IL-10 release in K562 cells. (C) Pentoxifylline (inhibitor of TNF-α) and (D) SP600125 (JNK inhibitor) restored the shikonin derivatives induced cell viability. The experiments were repeated three times, yielding similar results, and data are expressed as means ± SEM. *, p < 0.05; **, p < 0.01 significant difference compared with respective untreated control. | ||
3.6. Role of AgNPs in enhancing the cytotoxic effect of shikonin derivatives
AgNPs are known to induce cytotoxicity, DNA damage, and apoptosis.52 However, the jacalin-capped AgNPs prepared in this study were unable to induce significant cytotoxicity in K562 cells [Fig. S4†]. At the same time, the AgJAS/AgJBDS complexes induced a drastic increase in ROS-mediated apoptosis in K562 cells [Fig. 4], which suggested that JAgNPs act as a “Trojan horse” bypassing typical barriers and then releasing the drugs that damaged cell machinery through the mechanism of ROS.59 The internalization of the shikonin derivatives was confirmed by fluorescence microscopy (Fig. 9). The fluorescence image showed that significant amounts of AgJAS/AgJBDS were internalized in K562 cells, supporting the effectiveness of AgJAS/AgJBDS towards higher cell death. At the same time, AgJAS/AgJBDS was unable to internalize into non-tumorigenic HepG2 cells (Fig. S10†), indicating their high selectivity for the tumorigenic cells. | ||
| Fig. 9 Intracellular uptake of AgJAS (A–C) and AgJBDS (D–F) in K562 cells by fluorescence imaging. A required amount of cells was treated with rhodamine B labelled AgJAS/AgJBDS (100 nM) for 4 h. Intracellular uptake was examined using fluorescence microscope. | ||
As described in the results above, ROS was a mechanism of AS/BDS induced toxicity in K562 cells, but the enhancement of ROS by AS/BDS could not completely explain the enhanced cytotoxicity of AgJAS/AgJBDS. As noted from Fig. 4A and S6D,† JAgNPs induced small amounts of ROS generation, which may not be sufficient to induce cytotoxicity to K562 cells (refer Fig. S4†). However, Fig. 4B showed that NAC could completely reverse the ROS induced by AgJAS/AgJBDS and prevented the cells from undergoing apoptosis, which clearly suggests that ROS generated by AgNPs was also inhibited by NAC. Guo et al. showed that release of silver ions and the generation of ROS were suggested to play pivotal roles in the AgNPs induced cytotoxicity in AML cells, which can be hampered in the presence of NAC.60 NAC is well known for antioxidant activity. However, it can also serve as a silver ion chelator.61 In this study, indirect evidence for the release of silver ions from AgJAS/AgJBDS was obtained from the ROS inhibitor experiment [Fig. 4B] and it showed complete reversal of the cytotoxicity of shikonin derivatives, indicating that the release of silver ion from AgJAS/AgJBDs would be prevented in the presence of NAC. Liu et al. reported that the high rate of respiration in cancer cells could release silver ions from the AgNPs through intracellular oxidation.62 Taken together, we reasoned that chelation of silver ions and inhibition of the ROS by NAC leads to an increased cell viability in K562 even after exposing to AgJAS/AgJBDS [Fig. 4B]. Based on these data, we suggest that intracellular oxidation and ROS generated by AS/BDS promoted the oxidation of AgNPs and released silver ions from the AgNPs,60–62 which resulted in increased toxicity of AgJAS/AgJBDS that efficiently killed the K562 cells more than the plain shikonin derivatives [Fig. 3]. However, further studies are required to confirm the release of silver ions from the AgJAS/AgJBDS. The apoptotic events involved during the exposure of AgJAS/AgJBDS to K562 is shown in Fig. 10.
 | ||
| Fig. 10 Schematic representation of AgJAS/AgJBDS induced apoptosis. | ||
4. Conclusions
In this report, we demonstrated the interaction between jacalin and shikonin derivatives and used their interaction properties to enhance the cytotoxic potential of AS/BDS by loading into jacalin-capped AgNPs. JAgNPs containing AS/BDS showed a stronger anti-leukemia effect at lower dosage as compared to pure AS/BDS. The mechanistic study showed that the higher induction of ROS by AgNPs containing shikonin derivatives and release of silver ions from AgJAS/AgJBDS were important for their enhanced cytotoxicity effect. The data presented provides a novel approach to enhance drug efficacy for the treatment of diseases in the future.Acknowledgements
The Department of Science and Technology, Government of India (SB/FT/LS-217/2012) is acknowledged for the financial support. We thank central research facility (R&M/0021/SCBT-007/2012-13), SASTRA University for the infrastructure.References
- A. Jemal, F. Bray, M. Center, J. Ferlay, E. Ward and D. Forman, Cancer J. Clin., 2011, 61, 69 CrossRef PubMed.
- P. Vineis and C. P. Wild, Lancet, 2014, 383, 549 CrossRef.
- R. S. Magge and L. M. DeAngelis, Blood Rev., 2015, 29, 93 CrossRef CAS PubMed.
- K. Asić, Crit. Rev. Oncol. Hematol., 2015, pii S1040–8428, 30019 Search PubMed.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615 CrossRef CAS PubMed.
- T. G. Cotter, Nat. Rev. Cancer, 2009, 9, 501 CrossRef CAS PubMed.
- M. S. Baliga, S. Meera, L. K. Vaishnav, S. Rao and P. L. Palatty, Integr. Cancer Ther., 2013, 12, 455 CrossRef PubMed.
- M. R. Ven Murthy, P. K. Ranjekar, C. Ramassamy and M. Deshpande, Cent. Nerv. Syst. Agents Med. Chem., 2010, 10, 238 CrossRef CAS PubMed.
- X. Xiong, X. Li, Y. Zhang and J. Wang, BMJ Open, 2015, 5, e005355 CrossRef PubMed.
- X. Wang, Y. Feng, N. Wang, F. Cheung, H. Y. Tan, S. Zhong, C. Li and S. Kobayashi, BioMed Res. Int., 2014, 2014, 530342 Search PubMed.
- K. Sharma and R. Zafar, Pharmacogn. Rev., 2015, 9, 19 CrossRef CAS PubMed.
- S. Grabley and R. Thiericke, Adv. Biochem. Eng./Biotechnol., 1999, 64, 101 CrossRef CAS PubMed.
- D. Chaturvedi, A. Goswami, P. P. Saikia, N. C. Barua and P. G. Rao, Chem. Soc. Rev., 2010, 39, 435 RSC.
- S. Dall'Acqua, Curr. Top. Med. Chem., 2014, 14, 2272 CrossRef.
- X. Chen, L. Yang, J. J. Oppenheim and O. M. Z. Howard, Phytother. Res., 2002, 16, 199 CrossRef CAS PubMed.
- U. Sankawa, Y. Ebizuka, T. Miyazaki, Y. Isomura and H. Otsuka, Chem. Pharm. Bull., 1977, 25, 2392 CrossRef CAS PubMed.
- X. P. Guo, X. Y. Zhang and S. D. Zhang, Zhongxiyi Jiehe Zazhi, 1991, 11, 598 CAS.
- H. Wu, J. Xie, Q. Pan, B. Wang, D. Hu and X. Hu, PLoS One, 2013, 8, e52706 CAS.
- A. Arora, M. L. Gulrajani and D. Gupta, J. Nat. Prod. Resour., 2009, 8, 142 Search PubMed.
- G. Kumar and Y. K. Gupta, AYU, 2012, 33, 569 CrossRef PubMed.
- I. Ali, W. A. Wani, K. Saleem and A. Haque, Anticancer Agents Med. Chem., 2013, 13, 296 CrossRef CAS PubMed.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593 CAS.
- J. A. Ochoa-Alvarez, H. Krishnan, Y. Shen, N. K. Acharya, M. Han, D. E. McNulty, H. Hasegawa, T. Hyodo, T. Senga, J. G. Geng, M. Kosciuk, S. S. Shin, J. S. Goydos, D. Temiakov, R. G. Nagele and G. S. Goldberg, PLoS One, 2012, 7, e41845 CAS.
- M. A. Hassan, R. Rouf, E. Tiralongo, T. W. May and J. Tiralongo, Int. J. Mol. Sci., 2015, 16, 7802 CrossRef CAS PubMed.
- G. Poiroux, M. Pitié, R. Culerrier, B. Ségui, E. J. Van Damme, W. J. Peumans, J. Bernadou, T. Levade, P. Rougé, A. Barre and H. Benoist, PLoS One, 2011, 6, e23315 CAS.
- S. Sakuma, S. Yamashita, K. Hiwatari, R. M. Hoffman and W. Pham, Curr. Drug Discovery Technol., 2011, 8, 367 CrossRef CAS.
- P. Dubey, I. Matai, S. U. Kumar, A. Sachdev, B. Bhushan and P. Gopinath, Adv. Colloid Interface Sci., 2015, 221, 4 CrossRef CAS PubMed.
- M. V. Sastry, P. Banarjee, S. R. Patanjali, M. J. Swamy, G. V. Swarnalatha and A. Surolia, J. Biol. Chem., 1986, 261, 11726 CAS.
- A. A. Jeyaprakash, P. Geetha Rani, G. Banuprakash Reddy, S. Banumathi, C. Betzel, K. Sekar, A. Surolia and M. Vijayan, J. Mol. Biol., 2002, 23, 637 CrossRef.
- V. S. Marangoni, I. M. Paino and V. Zucolotto, Colloids Surf., B, 2013, 112, 380 CrossRef CAS PubMed.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 6158 CrossRef CAS PubMed.
- S. S. Komath, K. Bhanu, B. G. Maiya and M. J. Swamy, Biosci. Rep., 2000, 20, 265 CrossRef CAS PubMed.
- S. S. Komath, M. Kavitha and M. J. Swamy, Org. Biomol. Chem., 2006, 4, 973 CAS.
- G. Pandey, T. Fatma, S. M. Cowsik and S. S. Komath, J. Photochem. Photobiol., B, 2009, 97, 87 CrossRef CAS PubMed.
- K. B. Ayaz Ahmed, E. Reshma, M. Mariappan and V. Anbazhagan, Spectrochim. Acta, Part A, 2015, 137, 1292 CrossRef CAS PubMed.
- K. B. Ayaz Ahmed, A. S. Mohammed and A. Veerappan, Spectrochim. Acta, Part A, 2015, 145, 110 CrossRef CAS PubMed.
- T. Hongbo, L. Yanping, S. Min and W. Xiguang, Polym. J., 2012, 44, 211 CrossRef.
- S. Subramaniam, A. Palanisamy and A. Sivasubramanian, RSC Adv., 2015, 5, 6265 RSC.
- S. Subramaniam, N. Rajendran, S. B. Muralidharan, G. Subramaniam, R. Raju and A. Sivasubramanian, RSC Adv., 2015, 5, 77168 RSC.
- P. Padma, S. S. Komath, S. K. Nadimpalli and M. J. Swamy, Phytochemistry, 1999, 50, 363 CrossRef CAS.
- N. A. M. Sultan, B. G. Maiya and M. J. Swamy, Eur. J. Biochem., 2004, 271, 3274 CrossRef CAS PubMed.
- S. K. Dash, S. S. Dash, S. Chattopadhyay, T. Ghosh, S. Tripathy, S. Kar Mahapatra, B. G. Bag, D. Das and S. Roy, RSC Adv., 2015, 5, 24144 RSC.
- S. Kar Mahapatra, S. Bhattacharjee, S. P. Chakraborty, S. Majumdar and S. Roy, Int. Immunopharmacol., 2011, 11, 485 CrossRef PubMed.
- W. Han, J. Xie, L. Li, Z. Liu and X. Hu, Apoptosis, 2009, 14, 674 CrossRef CAS PubMed.
- V. P. Papageorgiou, A. N. Assimopoulou, E. A. Couladouros, D. Hepworth and K. C. Nicolaou, Angew. Chem., Int. Ed., 1999, 38, 270 CrossRef.
- S. K. Mahanta, M. V. K. Sastry and A. Surolia, Biochem. J., 1990, 265, 831 CrossRef CAS PubMed.
- E. Kelly, L. Coronado, L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef.
- T. Kitao and K. Hattori, Nature, 1997, 265, 81 CrossRef.
- S. Chattopadhyay, S. K. Dash, S. Tripathy, B. Das, S. K. Mahapatra, P. Pramanik and S. Roy, J. Appl. Toxicol., 2015, 35, 603 CrossRef CAS PubMed.
- S. Gurunathan, J. Raman, S. N. A. Malek, P. A. John and S. Vikineswary, Int. J. Nanomed., 2013, 8, 4399 Search PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195 CrossRef CAS PubMed.
- M. J. Piao, K. A. Kang, I. K. Lee, H. S. Kim, S. Kim, J. Y. Choi, J. Choi and J. W. Hyun, Toxicol. Lett., 2011, 201, 92 CrossRef CAS PubMed.
- R. K. Gara, V. K. Srivastava, S. Duggal, J. B. Bagga, M. L. B. Bhatt, S. Sanyal and D. Mishra, J. Biomed. Sci., 2015, 22, 26 CrossRef PubMed.
- P. C. Hsu, Y. T. Huang, M. L. Tsai, Y. J. Wang, J. K. Lin and M. H. Pan, J. Agric. Food Chem., 2004, 52, 6330 CrossRef CAS PubMed.
- B. Wiench, T. Eichhorn, M. Paulsen and T. Efferth, Evidence-Based Complementary Altern. Med., 2012, 726025 Search PubMed.
- L. Wang, F. Du and X. Wang, Cell, 2008, 133, 693 CrossRef CAS PubMed.
- A. E. Serrano, E. Menares-Castillo, M. Garrido-Tapia, C. H. Ribeiro, C. J. Hernández, A. Mendoza-Naranjo, M. Gatica-Andrades, R. Valenzuela-Diaz, R. Zúñiga, M. N. López, F. Salazar-Onfray, J. C. Aguillón and M. C. Molina, Immunol. Cell Biol., 2011, 89, 447 CrossRef CAS PubMed.
- X. Mao, C. R. Yu, W. H. Li and W. X. Li, Cell Res., 2008, 18, 879 CrossRef CAS PubMed.
- E. J. Park, J. Yi, Y. Kim, K. Choi and K. Park, Toxicol. In Vitro, 2010, 24, 872e8 Search PubMed.
- D. Guo, L. Zhu, Z. Huang, H. Zhou, Y. Ge, W. Ma, J. Wu, X. Zhang, X. Zhou, Y. Zhang, Y. Zhao and N. Gu, Biomaterials, 2013, 34, 7884 CrossRef CAS PubMed.
- X. Y. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-Kim and J. N. Meyer, Environ. Sci. Technol., 2012, 46, 1119 CrossRef CAS PubMed.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR of AS, BDS; binding curve, haemolytic activity, mitochondrial membrane potential, ROS generation, nuclear morphology. See DOI: 10.1039/c5ra27952f |
| This journal is © The Royal Society of Chemistry 2016 |