
Evaluation of in-tube solid-phase microextraction method for co-extraction of acidic, basic, and neutral drugs†
Abstract
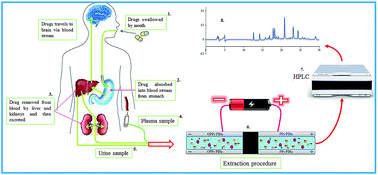
Simultaneous extraction of acidic, basic, and neutral drugs from different biological samples is a considerable and disputable concept in sample preparation strategies. In the present work, a new in-tube solid phase microextraction approach named electrochemically controlled double in-tube solid phase microextraction (EC-DIT-SPME) is introduced for simultaneous extraction of acidic, basic, and neutral drugs from different biological matrices. For this purpose, novel nanostructured coatings based on copolymer of polypyrrole and indole-2-carboxylic acid (PPy-co-PIca) as non-overoxidized and overoxidized forms were electrochemically synthesized on the inner surface of stain-less steel tubes. During the extraction procedure, the PPy-co-PIca and overoxidized PPy-co-PIca coated stain-less steel tubes were used as anode and cathode electrodes, respectively. Extraction of basic and acidic drugs were performed on the surface of overoxidized PPy-co-PIca and PPy-co-PIca, which are connected to the negative and positive potentials, respectively. With regard to the fact that neutral drugs can be absorbed in both electrodes, extraction of neutral drugs will be done in both of them. Satisfactory analytical figures of merit including linear dynamic range and limits of detection in the range of 0.15–500 ng mL−1 and 0.05–1.9 ng mL−1 and good extraction recoveries (38.5–78.6%) were obtained. Finally, the proposed method was successfully applied for simultaneous analysis of acidic, basic, and neutral drugs in some biological samples.
 Please wait while we load your content...
Please wait while we load your content...

