
In situ hydrothermal growth of a dual-ligand metal–organic framework film on a stainless steel fiber for solid-phase microextraction of polycyclic aromatic hydrocarbons in environmental water samples†
Shu-Hui Huo*a,
Jing Yua,
Yan-Yan Fub and
Peng-Xin Zhoua
aKey Laboratory of Eco-Environment-Related Polymer Materials of Ministry of Education, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, China. E-mail: huosh@nwnu.edu.cn
bSchool of Medical Imaging, Tianjin Medical University, Tianjin 300203, China
First published on 25th January 2016
Abstract
Effective enrichment and determination of polycyclic aromatic hydrocarbons (PAHs) in environmental aqueous solutions is still a major challenge because of their poor solubility and low concentration in the environmental matrix. Solid-phase microextraction (SPME) has been proven to be an effective technique for analyzing trace analytes from environmental samples. Herein, we report the fabrication of a dual-ligand metal–organic framework (MOF) bio-MOF-1-coated stainless steel fiber via an in situ growth approach for SPME of six PAHs in water samples. Such MOF-based SPME in combination with gas chromatography (GC) gave enhancement factors of 3104–5980, ranges of 10–100 μg L−1 for Nap, Ace, and Fle, 2.5–100 μg L−1 for Phen, 0.1–100 μg L−1 for FluA and Pyr, detection limits (S/N = 3) of 0.02–5.57 μg L−1, and quantitation limits (S/N = 10) of 0.14–17.2 μg L−1 for the studied PAHs. The recoveries obtained by spiking 10 μg L−1 PAHs in water samples ranged from 80% to 115%. The results showed that π–π interaction with biphenyldicarboxylate and π-complexation between aromatic rings and the pyrimidine in the pores of bio-MOF-1 play significant roles in the extraction of PAHs.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a widely distributed group of organic pollutants that are well known because of their toxic, mutagenic, and carcinogenic effects, as well as their tendency to bioaccumulate, especially the PAHs containing four or more aromatic rings.1 Effective enrichment and determination of PAHs in environmental aqueous solutions is a major challenge because of their low solubility and concentration in the matrix.2 Hence, various techniques for effective preconcentration and separation of PAHs at trace levels have received extraordinary attention.Solid-phase microextraction (SPME), which integrates sampling, analyte isolation, concentration and sample introduction into a single step, was proposed by Pawliszyn's group in the 1990s.3 SPME is considered to be an environment-friendly technique and is widely applied in the analysis of environmental,4 food,5 forensic,6 pharmaceutical7 and biological samples8 because of its merits, such as solvent-free nature of the process, simple operation and easy automation.9 Absorbing materials coated the fiber play a key role in SPME because analytes in the sample are directly extracted and concentrated in the fiber coating. Currently, several commercial fibers such as poly(dimethylsiloxane) (PDMS),10 poly(dimethylsiloxane)/divinylbenzene (PDMS/DVB),11 and polyacrylate (PA)12 are available for SPME. However, some drawbacks such as insufficient thermal or solvent instability and fragility of substrate are still present. To overcome these problems, development of novel sorbents for SPME has become of great importance and poses a significant challenge.
Metal–organic frameworks (MOFs) are a fascinating class of highly ordered porous materials that exist as infinite crystalline lattices with metal centers/clusters and organic linkers, and possess accessible cages, tunnels and modifiable pores.13 MOFs show great potential for applications in many fields, especially in gas storage14 and separation,15–20 adsorption,21,22 extraction,23–25 sensing,26 chiral discrimination27 and imaging.28,29 Unique characteristics such as large surface area,30 good thermal stability, uniform structured nanoscale cavities, and their tunable nature29,31 have also made MOFs attractive as highly efficient sorbents in SPME.
To date, MOFs such as MOF-199,4 ZIF-8,18,32 ZIF-90,33 MIL-53,34 MIL-88B,35 MIL-101,36 IRMOF-3 (ref. 37) and UIO-66 (ref. 38) have been shown to be promising as coatings for the SPME of diverse analytes including gaseous benzene homologues,4 n-alkanes,18 polycyclic aromatic hydrocarbons,34 polychlorinated biphenyls,35 and phenolic endocrine disruptors.33,38 The pioneering works were performed in headspace mode, and the MOFs utilized as coatings for SPME were bridged with a single ligand. When required, SPME fibers were immersed directly in environmental aqueous samples and equilibrated in situ. Moreover, the exploration of novel coatings for the SPME requires novel materials with different structure, composition, adsorption properties than former materials. Herein, we report the fabrication of a dual-ligand MOF (bio-MOF-1)-coated stainless steel fiber via in situ growth approach for the SPME of PAHs in water samples. Bio-MOF-1, Zn8(ad)4(BPDC)6O·2Me2NH2, reported by Rosi's group,39 shows a three dimensional permanently porous MOF with infinite zinc–adeninate columnar secondary building units (SBU), which are interconnected via biphenyldicarboxylate linkers.40 Bio-MOF-1 is an appealing candidate for coatings for SPME because it has permanent microporosity, excellent high thermal and chemical stability, high surface areas, and exceptional adsorption capacity due to the presence of π-electrons in aromatic rings and the basic bio-molecule building units.41 To the best of our knowledge, few studies on the fabrication of metal–adeninate biometal organic frameworks MOFs bonded on stainless steel fiber for SPME have been reported so far.
The bio-MOF-1-coated fiber was obtained via in situ hydrothermal reaction of Zn(II), adeninate and biphenyldicarboxylate. The bio-MOF-1-coated fiber obtained was characterized by scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), X-ray powder diffractometry (XRD), and thermal gravimetric analysis (TGA) and applied to the efficient enrichment of six PAHs from water samples. Effects of extraction temperature and time, pH, ionic strength, agitation speed and desorption time were studied in detail. The developed method was then applied to the determination of PAHs in environmental water samples.
2. Experimental
2.1 Chemicals and materials
All reagents used were at least of analytical grade. 4,4′-Biphenyl dicarboxylic acid (BPDC), zinc acetate dihydrate and adenine (Aladdin, Shanghai, China) were used to prepare bio-MOF-1. Nitric acid (65%) and hydrofluoric acid (40.0%) were purchased from Tianjin Standard Science and Technology Co., Ltd. (Tianjin, China). Acetonitrile, anhydrous ethanol, and N,N-dimethylformamide (DMF) were obtained from Concord Technology (Tianjin, China). Stainless steel wire was obtained from Shanghai Gaoge Industrial and Trade Co., Ltd. (Shanghai, China). Ultrapure water (18.2 MΩ cm) was obtained from a WaterPro Water Purification System (Labconco Corporation, Kansas City, MO, USA). Certified individual standards of naphthalene (Nap), acenaphthene (Ace), fluorene (Fle), phenanthrene (Phen), fluoranthene (FluA), and pyrene (Pyr) were purchased from Aldrich (St. Louis, MO, USA). Individual stock solutions of the PAHs were prepared in acetonitrile. The working standard solution was prepared by combining aliquots of each individual stock solution and diluting to obtain the desired concentration. The volume portion of acetonitrile in any working solution was less than 1%. The solutions mentioned above were kept at 4 °C in the dark. Caution: Take care to avoid direct contact with all of the analytes studied and prepare all of the solutions in a well-ventilated hood because of their high toxicity.2.2 Apparatus
The thermogravimetric analysis (TGA) was performed on a PTC-10A thermal gravimetric analyzer (Rigaku, Japan) from room temperature to 800 °C at a ramp rate of 10 °C min−1. The X-ray diffraction (XRD) patterns were performed on a diffractometer (D/max-2500, Rigaku, Japan) using Cu Kα radiation (λ = 1.5418 Å) over the angular range from 3° to 80°. The fiber morphology was characterized using a field emission scanning electron microscope (Ultra Plus, Carl Zeiss, Germany). The chemical composition of the fiber surface was investigated using EDS (Oxford). A Thermo Trace Ultra gas chromatograph (GC) equipped with a flame ionic detector (FID) was employed for all experiments. A GC capillary column (SE-54, 30 m length × 0.53 mm i.d. × 1.0 μm) was purchased from Lanzhou Institute of Chemical Physics (Lanzhou, China). The column temperature was initially maintained at 160 °C for 3 min and then heated at 15 °C min−1 to the final temperature of 260 °C, where it was held for 5 min. The injector and detector temperatures were set at 270 °C and 300 °C, respectively. High-purity nitrogen (99.99%, BOC Gases Co. Ltd., Tianjin, China) was used as the carrier gas at a flow rate of 40 mL min−1. Hydrogen and air were maintained at flow rates of 50 and 450 mL min−1, respectively. Splitless injections were used throughout. A model 85-1 stir plate (Jintan Instruments Co. Ltd., Jintan, Jiangsu) and a Teflon-coated magnetic stir bar (9.9 mm × 5.9 mm × 5 mm) were used for agitation.2.3 In situ growth of bio-MOF-1 film on SPME fiber
Stainless steel wires with a length of 6.0 cm were used to fabricate the SPME fibers. Half (3.0 cm in length) of the stainless steel wire was etched by hydrofluoric acid to generate a rough surface, washed gently with ultrapure water, and dried in air. As shown in Scheme 1, for in situ hydrothermal growth of the bio-MOF-1 films onto the hydrofluoric acid-etched part of the stainless steel wire, adenine (0.063 mmol), BPDC (0.125 mmol), zinc acetate dihydrate (0.19 mmol), nitric acid (1 mmol), DMF (13.5 mL), and water (1 mL) were added to a 30.0 mL Teflon vial. The hydrofluoric acid-etched stainless steel wire was carefully immersed in the solution. The vial was sealed and placed in a steel autoclave at 130 °C for 24 h. After cooling to room temperature, the bio-MOF-1-coated fiber was taken out of the Teflon liner with great care, washed gently with DMF and ultrapure water sequentially, and dried at 120 °C under N2. Thus, the resulting bio-MOF-1-coated fiber was obtained.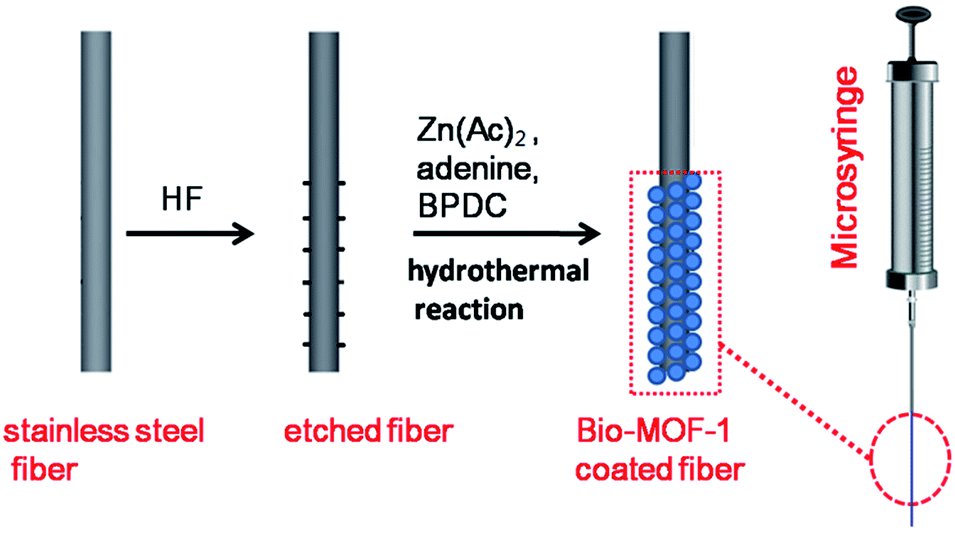 | ||
| Scheme 1 Schematic demonstration for fabricating a bio-MOF-1 coated SPME fiber. | ||
The bio-MOF-1-coated SPME fiber was firmly fixed in place by inserting the uncoated part of the stainless steel wire into a stainless steel capillary, which was then loaded into a 5 μL GC microsyringe by replacing the plunger of the microsyringe. The SPME fiber could be detached from the stainless steel capillary when necessary. Before use, the fiber was conditioned in the GC injector at 270 °C for 30 min under N2.
2.4 Sample pretreatment
River water and waste water samples were collected locally. The water samples were filtered with 0.45 μm filter membranes and stored in clean glass bottles which were successively washed with detergents, water, methanol, and ultrapure water, and dried before use. The pH value of water samples was adjusted with 0.1 mol L−1 HCl and ammonia prior to analysis. The water samples were analyzed immediately after sampling.2.5 Analytical procedures
To perform the extraction, 20 mL of either the aqueous standard solution or a sample solution was placed in a 25 mL glass vial. The vial was tightly capped with a butyl rubber stopper wrapped with polytetrafluoroethylene sealing tape after introducing a Teflon-coated magnetic stir bar. The needle of the SPME device was passed through the septum, and the bio-MOF-1-coated fiber was extended until immersed in the solution. The extraction was carried out at 50 °C while stirring at 1200 rpm. After 30 min of extraction, the fiber was removed from the vial and immediately transferred to the GC injection port for thermal desorption at 270 °C for 5 min. The fiber was conditioned at 270 °C for 10 min between two extractions.3. Results and discussion
3.1 Characterization of bio-MOF-1-coated fiber
In the present work, the bio-MOF-1 film was grown hydrothermally in situ on the surface of the stainless steel fiber (Scheme 1). The surface morphology and chemical composition of the etched stainless steel fiber before and after in situ-fabrication were analyzed in order to determine the reasons for the affinity of bio-MOF-1 film on etched stainless steel fiber to PAHs (Fig. 1). Compared to the rough surface on etched stainless steel fiber (Fig. 1A), the as-fabricated fiber shows that bio-MOF-1 deposits are uniformly spaced on the surface of the stainless steel fiber with relatively uniform size and structure (Fig. 1B). Typical rod-shaped crystals (approximately 10–12 μm in length) on the surface of stainless steel fiber as confirmed by the formation of crystalline bio-MOF-1, epitaxially oriented to form a homogenous film (Fig. 1C). Elemental mapping by EDS analysis of the bio-MOF-1 fiber at an accelerating voltage of 10 kV revealed that the elements Fe, Cr, C, F and Zn are homogeneously distributed throughout the whole fiber (Fig. S1†). On the substrate, chemical bonding also occurred due to abundance of an oxygen or metal-containing group such as Fe2O3, FeF3, Cr2O3, and CrF2 on the surface of the etched stainless steel wire,42 which is beneficial for both adhesion and orientation of bio-MOF-1 crystallites via multiple BPDC linkers interconnected with zinc–adeninate columnar secondary building units. EDS analysis revealed that the elements C, O, N and Zn are homogeneously distributed throughout the bio-MOF-1 film (Fig. 1D–G). The weight percentages of C, O, N and Zn on the surface of rod-shaped bio-MOF-1 crystallites were 56.8%, 15.9%, 11.67% and 15.65%, respectively. From the point of view of porous nature, the crystal may be formed by the adsorption of zinc salts and organic linkers (adenine and BPDC) precursors of the zinc–adeninate columnar secondary building units in the pore for the oriented growing bio-MOF-1 crystallites. | ||
| Fig. 1 SEM images of (A) stainless steel wire; (B) and (C) bio-MOF-1 SPME fiber. EDS elemental mapping images for bio-MOF-1 SPME fiber (D)–(G). | ||
A typical XRD pattern of the powder scraped from the bio-MOF-1 fiber is in good agreement with the simulated one, further indicating the successful fabrication of the bio-MOF-1 layer on the fiber (Fig. S2A†). The thermal resistance of the fiber coating is very important for SPME in combination with GC. The thermal stability of bio-MOF-1 layer was evaluated by TGA. The weight loss of approximately 20.7% near 290 °C was attributed to guest molecules in the pore. After aging at higher temperature, there was no obvious weight loss, indicating that the thermostability of bio-MOF-1 is sufficient for the SPME application (Fig. S2B†). The Brunauer–Emmett–Teller (BET) surface area of bio-MOF-1 scraped from the fibers was 1083 m2 g−1, which ensures the excellent adsorption ability and high analyte capacity of the fibers.
3.2 Optimization of conditions for SPME of PAHs
Potential factors affecting the SPME of PAHs on bio-MOF-1 were investigated and optimized, including extraction temperature, extraction time, ionic strength, pH, agitation speed, and desorption time.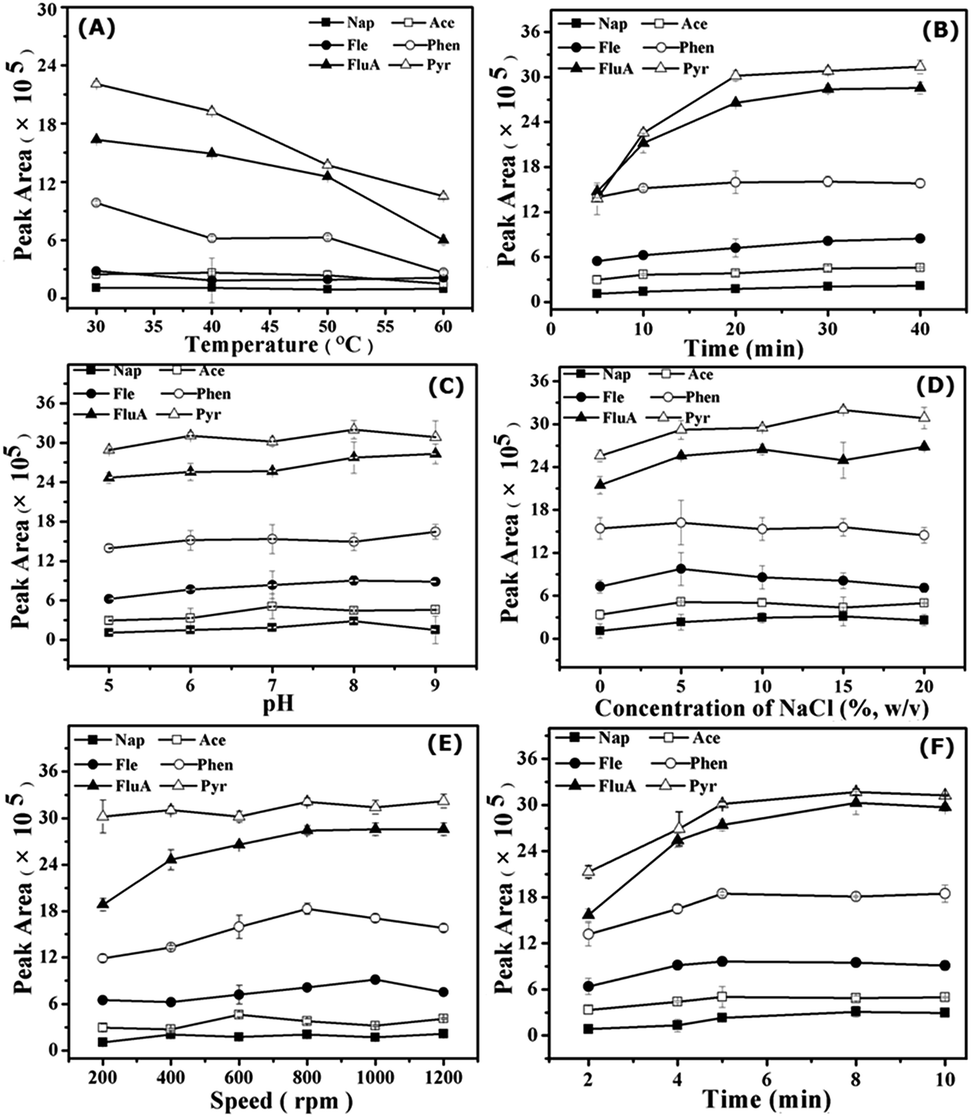 | ||
| Fig. 2 Factors affecting the extraction efficiency for 50 μg L−1 PAHs. Effect of extraction temperature (A), time (B), pH (C), ionic strength (D), agitation speed (E) and desorption time (F) on the extraction efficiency. Error bars represent standard deviations calculated from triplicate extractions. | ||
However, solution pH could affect the stability and charge on the surface of bio-MOF-1. Hence, pH 8.0 was chosen as a compromise in the following extractions. The results show that the interaction between bio-MOF-1 and PAH molecules made a principal contribution to the extraction. The PAHs with highly delocalized π electrons allow π–π interaction with the biphenyldicarboxylate molecules in the framework of bio-MOF-1, and the π-complexation between aromatic rings and the pyrimidine in the pores of bio-MOF-1.
3.3 Analytical figures of merit
The analytical figures of merit for the determination of PAHs by the developed SPME method are summarized in Table 1. With the consumption of 20.0 mL of sample solution, the enrichment factors (EFs) of the PAHs (defined as the ratio of sensitivity after SPME to that obtained by direct injection of 1.0 μL of standard solution42) ranged from 3104 to 5980. The linearity of the developed SPME method was tested for the PAHs concentration range from 0.1 to 100 μg L−1 with a correlation coefficient (R2) between 0.980 and 0.999. The detection limits (DLs) (S/N = 3) and quantitation limits (QLs) (S/N = 10) for the PAHs are in the range of 0.02 to 5.57 μg L−1 and 0.14–17.2 μg L−1, respectively. The precision (relative standard deviations, RSDs) for six replicate determinations of the PAHs at 10 μg L−1 using a single bio-MOF-1-coated fiber ranges from 2.3% to 7.1%. The fiber-to-fiber reproducibility (RSDs, n = 3) for three parallel bio-MOF-1-coated fibers varies from 4.3% to 9.3%. Compared with previous SPME methods from the literature that use various sorbents for GC determination of PAHs, the present method gave lower DLs for the PAHs studied (Table 2). The performance of coatings in consecutive adsorption/desorption cycles is an important factors in SPME technique. To determine uptake capacity and reproducibility of bio-MOF-1-coated fiber, the six PAHs (50 μg L−1) were extracted with single fiber for ten replicates each day. The experiment was repeated for 6 days to complete 60 cycles of SPME with the same bio-MOF-1-coated fiber. The results show that the bio-MOF-1-coated fiber allowed 30 replicate extractions for six PAHs with relative standard deviations of less than 10.2% (see Fig. S3†).| Analyte | Linear range (μg L−1) | R2 | DLs (μg L−1) | QLs (μg L−1) | EFs (mean ± s, n = 3) | Precision for one fiber (%) | Fiber-to-fiber reproducibility (%) |
|---|---|---|---|---|---|---|---|
| Nap | 10–100 | 0.982 | 1.39 | 7.72 | 3104 ± 252 | 7.1 | 5.2 |
| Ace | 10–100 | 0.997 | 2.31 | 8.76 | 3375 ± 193 | 5.9 | 4.3 |
| Fle | 10–100 | 0.980 | 1.77 | 6.34 | 3445 ± 125 | 4.3 | 8.9 |
| Phen | 2.5–100 | 0.992 | 0.52 | 1.62 | 3878 ± 102 | 4.7 | 6.9 |
| FluA | 0.1–100 | 0.998 | 0.03 | 0.10 | 5140 ± 281 | 2.3 | 6.5 |
| Pyr | 0.1–100 | 0.999 | 0.02 | 0.08 | 5980 ± 245 | 5.5 | 9.3 |
| Coatings | Technique | Sample volume (mL) | EFs | LODs (μg L−1) | Ref. |
|---|---|---|---|---|---|
| a Not reported. | |||||
| IRMOF-3@ILs/PDMS | SPME-GC-MS | 20 | 4663–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 114 114 |
0.012–0.015 | 37 |
| PEDOT/GO composite | SPME-GC-FID | 9 | —a | 0.05–0.13 | 46 |
| PDMS/DVB commercial fiber | SPME-GC-FID | 20 | —a | 0.03–1.0 | 47 |
| Stainless steel wire | SPME-GC-FID | 10 | 2541–3981 | 0.24–0.63 | 42 |
| Bio-MOF-1 | SPME-GC-FID | 20 | 3140–5980 | 0.02–5.57 | This work |
3.4 Comparison of the bio-MOF-1-coated fiber with commercial PDMS fiber
In order to evaluate the performance of self-made bio-MOF-1-coated fiber toward PAHs, commercial PDMS fiber was selected for comparison under the same condition (Fig. 3). The bio-MOF-1-coated fiber gave much larger EFs (3140–5980) for PAHs than commercial PDMS fiber (EFs = 391–1482). The above results revealed the great potential of bio-MOF-1 coated fiber as an advanced fiber for the SPME of PAHs.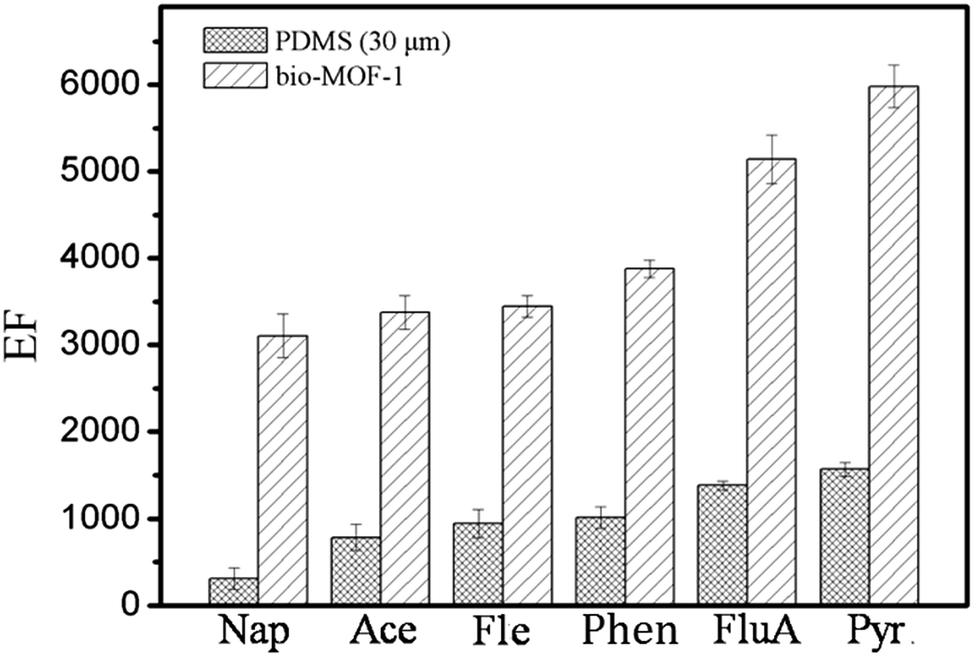 | ||
| Fig. 3 Comparison of the bio-MOF-1 coated fiber with commercial PDMS fiber for the SPME of PAHs. | ||
3.5 Sample analysis
The developed bio-MOF-1-coated fiber was applied to the SPME of PAHs analysis in local water samples. Furthermore, addition of certified PAHs was employed to identify the PAHs in real water sample and examine possible matrix effect. The analytical results were summarized in Table 3. FluA and Pyr were determined to be 1.7 and 1.1 μg L−1 in the wastewater II sample. No PAHs were detected in the water and wastewater I samples. The recoveries obtained for spiked 10 μg L−1 PAHs in the samples ranged from 90.6% to 106.3%. Chromatograms of PAHs in wastewater sample and spiked sample are shown in Fig. 4.| River water | Wastewater I | Wastewater II | ||||
|---|---|---|---|---|---|---|
| No spiking | Spiked with 10 μg L−1 | No spiking | Spiked with 10 μg L−1 | No spiking | Spiked with 10 μg L−1 | |
| a Not detected or lower than LOD. | ||||||
| Nap | nda | 9.7 ± 0.6 | nd | 9.7 ± 0.3 | nd | 10.5 ± 0.3 |
| Ace | nd | 9.0 ± 0.8 | nd | 9.8 ± 0.2 | nd | 9.4 ± 0.5 |
| Fle | nd | 8.7 ± 0.3 | nd | 9.9 ± 0.3 | nd | 9.1 ± 0.5 |
| Phen | nd | 9.1 ± 0.1 | nd | 10.3 ± 0.3 | nd | 9.7 ± 0.7 |
| FluA | nd | 9.7 ± 0.5 | nd | 9.3.± 0.5 | 1.7 | 12.3 ± 0.2 |
| Pyr | nd | 8.8 ± 0.4 | nd | 9.0 ± 0.6 | 1.1 | 11.3 ± 0.3 |
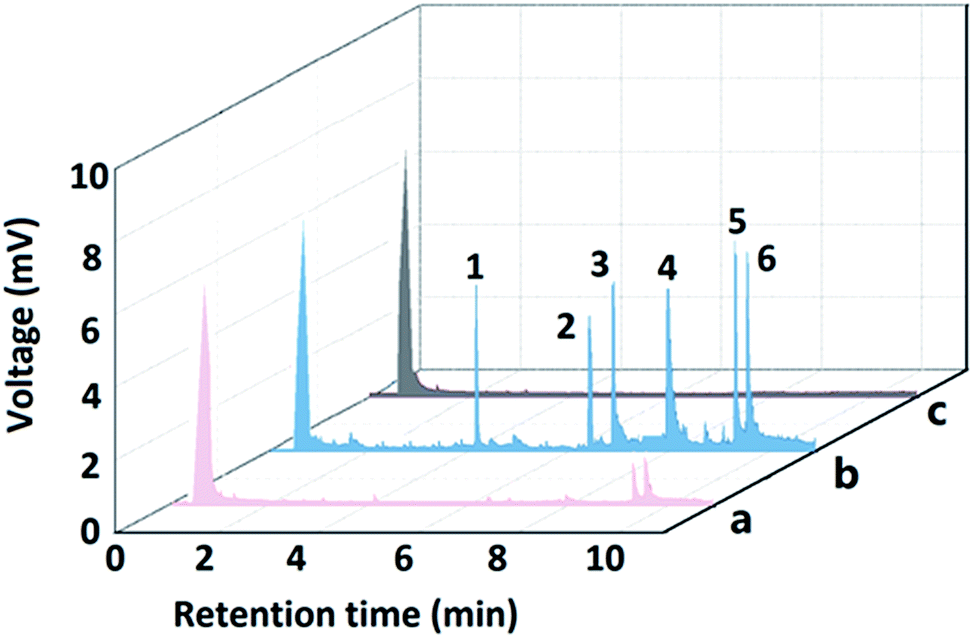 | ||
| Fig. 4 Chromatograms of the PAHs obtained by the developed method: (a) wastewater sample II; (b) spiked wastewater sample II with 50 μg L−1 of standard PAHs. (c) River sample. Peak identify: 1, Nap; 2, Ace; 3, Fle; 4, Phen; 5, FluA; 6, Pyr. | ||
4. Conclusions
The dual-ligand bio-MOF-1-coated stainless steel fiber was fabricated via an in situ growth approach and was applied as a novel fiber for the SPME of PAHs in water samples with large enhancement factors, low detection limits, good thermal and solvent stability and reproducibility. The results also reveal the great potential of MOF-based coatings for SPME of trace targets in complex environmental matrices.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21305112), China Postdoctoral Science Foundation (Grants 2015M571270), and Scientific and Technical Innovation Project of Northwest Normal University (Grants LKQN-12-3).References
- M. S. J. Chorazy, M. Strozyk and B. Cimander, Environ. Health Perspect., 1994, 102, 61 CrossRef CAS PubMed.
- L. Wolska, K. Galer, T. Gorecki and J. Namiesnik, Talanta, 1999, 50, 985 CrossRef CAS PubMed.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145 CrossRef CAS.
- X. Y. Cui, Z. Y. Gu, D. Q. Jiang, Y. Li, H. F. Wang and X. P. Yan, Anal. Chem., 2009, 81, 9771 CrossRef CAS PubMed.
- F. S. Mirnaghi, F. Mousavi, S. M. Rocha and J. Pawliszyn, J. Chromatogr. A, 2013, 1276, 12 CrossRef CAS PubMed.
- Y. He, J. Pohl, R. Engel, L. Rothman and M. Thomas, J. Chromatogr. A, 2009, 1216, 4824 CrossRef CAS PubMed.
- J. Xu, S. Huang, R. Wu, R. Jiang, F. Zhu, J. Wang and G. F. Ouyang, Anal. Chem., 2015, 87, 3453 CrossRef CAS PubMed.
- G. F. Ouyang, D. Vuckovic and J. Pawliszyn, Chem. Rev., 2011, 111, 2784 CrossRef CAS PubMed.
- G. A. Gomez-Rios and J. Pawliszyn, Angew. Chem., Int. Ed., 2014, 53, 14503 CrossRef CAS PubMed.
- E. Baltussen, P. Sandra, F. David, H. G. Janssen and C. Cramers, Anal. Chem., 1999, 71, 5213 CrossRef CAS.
- T. Gorecki, X. M. Yu and J. Pawliszyn, Analyst, 1999, 124, 643 RSC.
- C. L. Rainey, D. E. Bors and J. V. Goodpaster, Anal. Chem., 2014, 86, 11319 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS PubMed.
- C. X. Yang, Y. J. Chen, H. F. Wang and X. P. Yan, Chem.–Eur. J., 2011, 17, 11734 CrossRef CAS PubMed.
- Z. Y. Gu, C. X. Yang, N. Chang and X. P. Yan, Acc. Chem. Res., 2012, 45, 734 CrossRef CAS PubMed.
- C. X. Yang and X. P. Yan, Anal. Chem., 2011, 83, 7144 CrossRef CAS PubMed.
- N. Chang, Z. Y. Gu and X. P. Yan, J. Am. Chem. Soc., 2010, 132, 13645 CrossRef CAS PubMed.
- F. Yang, C. X. Yang and X. P. Yan, Talanta, 2015, 137, 136 CrossRef CAS PubMed.
- Y. Y. Wu, C. X. Yang and X. P. Yan, Analyst, 2015, 140, 3107 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- S. H. Huo and X. P. Yan, J. Mater. Chem., 2012, 22, 7449 RSC.
- S. H. Huo and X. P. Yan, Analyst, 2012, 137, 3445 RSC.
- Y. Y. Zhou, X. P. Yan, K. N. Kim, S. W. Wang and M. G. Liu, J. Chromatogr. A, 2006, 1116, 172 CrossRef CAS PubMed.
- D. Y. Lyu, C. X. Yang and X. P. Yan, J. Chromatogr. A, 2015, 1393, 1 CrossRef CAS PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS PubMed.
- H. J. Duan, C. X. Yang and X. P. Yan, RSC Adv., 2015, 5, 30577 RSC.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. G. Xie, S. Tran and W. B. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS PubMed.
- D. Y. Hong, Y. K. Hwang, C. Serre, G. Ferey and J. S. Chang, Adv. Funct. Mater., 2009, 19, 1537 CrossRef CAS.
- J. Zhang, W. Zhang, T. Bao and Z. Chen, J. Chromatogr. A, 2015, 1388, 9 CrossRef CAS PubMed.
- L. Q. Yu and X. P. Yan, Chem. Commun., 2013, 49, 2142 RSC.
- X. F. Chen, H. Zang, X. Wang, J. G. Cheng, R. S. Zhao, C. G. Cheng and X. Q. Lu, Analyst, 2012, 137, 5411 RSC.
- Y. Y. Wu, C. X. Yang and X. P. Yan, J. Chromatogr. A, 2014, 1334, 1 CrossRef CAS PubMed.
- L. Xie, S. Liu, Z. Han, R. Jiang, H. Liu, F. Zhu, F. Zeng, C. Su and G. F. Ouyang, Anal. Chim. Acta, 2015, 853, 303 CrossRef CAS PubMed.
- J. Zheng, S. Li, Y. Wang, L. Li, C. Su, H. Liu, F. Zhu, R. Jiang and G. F. Ouyang, Anal. Chim. Acta, 2014, 829, 22 CrossRef CAS PubMed.
- H. B. Shang, C. X. Yang and X. P. Yan, J. Chromatogr. A, 2014, 1357, 165 CrossRef CAS PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376 CrossRef CAS PubMed.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. P. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220 CrossRef CAS PubMed.
- J. A. Bohrman and M. A. Carreon, Chem. Commun., 2012, 48, 5130 RSC.
- H. L. Xu, Y. Li, D. Q. Jiang and X. P. Yan, Anal. Chem., 2009, 81, 4971 CrossRef CAS PubMed.
- H. Fu and D. Zhu, Anal. Chem., 2012, 84, 2366 CrossRef CAS PubMed.
- J. Pawliszyn, Anal. Chem., 2003, 75, 2543 CrossRef CAS PubMed.
- J. Ai, Anal. Chem., 1997, 69, 1230 CrossRef CAS.
- M. H. Banitaba, S. S. H. Davarani and S. K. Movahed, J. Chromatogr. A, 2014, 1325, 23 CrossRef CAS PubMed.
- M. C. Wei and J. F. Jen, Talanta, 2007, 72, 1269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26656d |
| This journal is © The Royal Society of Chemistry 2016 |