
High-contrast mechanochromism and polymorphism-dependent fluorescence of difluoroboron β-diketonate complexes based on the effects of AIEE and halogen†
Abstract
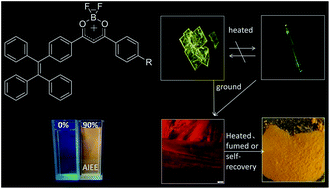
Tetraphenylethylene (TPE) is known for its aggregation-induced emission enhancement (AIEE) effect due to the restriction of intramolecular rotation. Some halogen-substituted fluorophores show interesting luminescent phenomena via C–H⋯halogen interactions. By combining TPE units, a halogen with a difluoroboron β-diketonate fluorophore, three complexes, 2a, 2b and 2c, were designed and synthesized. All of them exhibited intramolecular charge transfer, high fluorescence quantum yields, AIEE characteristics and high-contrast mechanochromism. By either grinding-fuming or grinding–annealing cycles, the pristine state or the original emission color of the three complexes can be almost recovered reversibly. In particular, 2c exhibits spontaneous reversibility after grinding at room temperature. Owing to the effect of halogen (Cl, Br), 2b and 2c show polymorphism-dependent fluorescence (yellow plate-like crystals and green needlelike crystals) and higher contrast than 2a before/after grinding. The maximum fluorescence emission wavelength of 2b and 2c shifts bathochromically 70 nm and 59 nm after grinding, respectively. To reveal the mechanochromic mechanism, a series of tests including powder XRD, fluorescence lifetime, DSC, and solid state UV-visible absorbance were completed. The results indicate that mechanochromism of the complexes can be correlated with the transformation between the amorphous and the crystalline states and metastable state formation after grinding, rather than molecular planarization, excimer or exciplex formation. To the best of our knowledge, this is the first example of no alkyl- or alkoxy-substituted difluoroboron β-diketonate complexes that exhibit reversible mechanochromic luminescence.
 Please wait while we load your content...
Please wait while we load your content...

