DOI:
10.1039/C5RA27817A
(Paper)
RSC Adv., 2016,
6, 22383-22388
A kinetic model for diffusion and chemical reaction of silicon anode lithiation in lithium ion batteries
Received
27th December 2015
, Accepted 11th February 2016
First published on 12th February 2016
Abstract
In this paper, a kinetic model is proposed that combines lithium ion diffusion through a lithiated phase with chemical reaction at the interface between lithiated amorphous and crystalline silicon. It is found out that a dimensionless parameter, relating the concentration distribution of lithium ions to the movement velocity of phase interface, can be used to describe the lithiation process. Based on the stress distributions and lithium ion diffusion profiles calculated by an elastic and perfectly plastic model, it is shown that, as lithiation proceeds, the hoop stress that changes from initial compression to tension in the surface layer of silicon particles may lead to surface cracking.
1. Introduction
Lithium ion batteries (LIBs) have been one of the most dominated power sources for renewable energy storage, electric vehicles and portable electronics.1–3 To obtain LIBs with a higher energy density, silicon is a promising anode material due to its high theoretical specific capacity (e.g., 4200 mA h g−1 for Li22Si5).4–6 During the lithium and delithium process, however, Si experiences a massive volume change (∼400%) and phase transition, which can result in diffusion-induced stress.7 Further, the stress can cause fragmentation, disintegration, fracture and debonding between current collectors and active materials.8,9
Extensive efforts have been made over the last decade to better understand lithiation degeneration.2,10 Based on previous research on bulk materials, the concentration distribution of Li ions is controlled by their diffusion in silicon.11–13 Recently, it was shown, however, that the diffusion-induced stress nanostructures such as nanotubes,14 hollow nanoparticles,15 and nanowires,16 can be alleviated through structure optimization and geometric restriction, which can relatively improve the cycle life compared to their bulk counterparts. In particular, a novel phenomenon occurs, where a lithiated amorphous LixSi phase was separated from the c-Si phase by a sharp phase boundary.16–20 This explains that with a decrease in size, the interfacial chemical reaction is more significant than the Li ion diffusion in silicon.21–23 Thus, it is necessary to elucidate the deformation–degeneration mechanism that is related to the lithiation phase transition, diffusion-induced stress and fracture.
To describe the coexistence of Li-poor and Li-rich phases, diffusivity was usually assumed to be nonlinearly dependent on the concentration of Li ions.9 Then, the Li ion concentration distribution and diffusion-induced stress can be obtained in such a two-phase microstructure. Furthermore, anisotropic swelling and fracture of silicon nanowires during lithiation were studied by a modified model.24,25 As is known, the phase field method can be applied to investigate multi-field problems such as the coupling of electrochemistry and mechanics.26,27 To clarify the driving force in a lithiated process, Zhao et al.28 assumed that the moving velocity of the interface was limited by the reaction of lithium and silicon rather than the diffusion of Li ions through the amorphous phase, and an analytical solution considering the chemical reaction and plastic deformation was obtained for a spherical particle. A kinetic plane model was suggested by introducing the redox reaction at the electrolyte/lithiated silicon interface, Li ion diffusion through the lithiated phase, and chemical reaction at the lithiated and crystalline Si interface.23
During lithiation, a large volume deformation may change silicon from a crystal to an amorphous phase and the latter can sustain inelastic deformation in the lithiated stage.29,30 A few theoretical models that couple the ion diffusion and stress have been applied to investigate the stress generation and fracture in crystalline silicon, in which the effects of composition-dependent modulus, nonlinear, inelastic and finite deformation, and boundary constraint were taken into account.11,12,31 Further, a strong size dependence of fracture was discovered, which means that there is a critical diameter, above which particles initially crack on surfaces and then fracture due to lithiation-induced swelling.9,24,32–36 In this paper, a kinetic model is established to consider the chemical reaction and Li ion diffusion through the lithiated phase in a small scale.
The paper is organized as follows. In Section 2, the kinetic model is presented based on several reasonable assumptions, and then analytic solutions on the Li ion concentration distribution and the moving velocity of phase interface are obtained. Further, a general mechanics model is proposed by means of the elastic–plastic deformation theory. In Section 3, the Li ion profile and the evolution of lithiated shell thickness and diffusion-induced stress field are discussed. Finally, the concluding remarks are given in Section 4.
2. Kinetic model
2.1. Model of interface-reaction controlled diffusion
As shown in Fig. 1, the crystalline core (c-Si) is surrounded by an amorphous shell (a-LixSi) in a core–shell Si nanowire. At first, the electrochemical charge-transfer reaction at electrode–electrolyte interfaces causes Li ions to be reduced with the intercalation of Li in electrode materials. The electrochemical reaction can be represented as| | |
Li (electrode surface) ↔ Li+ (electrolyte) + e− (electrode surface).
| (1) |
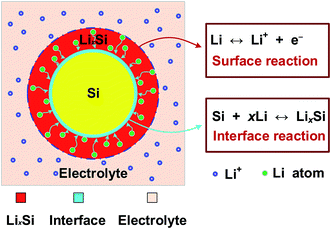 |
| | Fig. 1 Illustration of the kinetic model for a single particle of lithium–silicon anode. | |
Then, Li atoms diffuse through lithiated silicon, and react with crystalline silicon to form an amorphous phase at the reaction front, that is
Obviously, migration of Li ions in electrolyte is controlled by the Li diffusion through the LixSi phase and the reaction of Li and Si at interface between LixSi and Si. As is known, the amorphous phase of lithiated silicon is Li3.75Si at room temperature.9 Among all the possible LixSi compounds during electrochemical reactions, the compound with the maximum Li concentration is Li4.4Si. Thus, x is assumed to be between 3.75 and 4.4. To simplify the model, the solid electrolyte interphase (SEI) film deformation is neglected, though it may have a significant influence on the fracture of the active materials and degeneration of electrochemistry of LIBs.6,37–42
Here, it is worth noting that several assumptions are introduced as follows: (i) the Li diffusion process in lithiated silicon is regarded as a steady state. (ii) Li atoms diffusion occurs at lithiated silicon and the reaction appears at the front of a moving surface. (iii) The Li concentration on the particle surface reaches the maximum value initially, so the amorphous phase is considered as Li4.4Si. (iv) The Li concentration of a particle linearly decreases with the thickness of lithiated silicon. (v) The diffusion coefficient of Li in lithiated silicon is regarded as a constant.
In an axisymmetric model, the diffusion of solute atoms can be described by the classical Fick’s second law in a cylindrical coordinate system (r, φ, θ), that is
| |
 | (3) |
where
C is the ion concentration,
t is the reaction time, and
D is the diffusion coefficient of ions in electrode. According to assumption (i), we have
| |
 | (4) |
and its general solution is
| |
C = A + B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnr, lnr,
| (5) |
where
A and
B are the coefficients that can be determined by the boundary conditions.
The Li ion concentration and flux on surface are described as Cb and Jb, respectively, and their boundary conditions are
| |
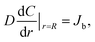 | (6) |
and
where
R is the radius of a nanowire. Thus,
eqn (5) can be rewritten as
| |
 | (8) |
At the interface between amorphous LixSi and crystalline Si phases, the diffusion interface reaction occurs, as shown in Fig. 1. The reaction is driven by the excess Li in lithiated silicon and its reaction rate controls the Li flux across the interface. The corresponding Li flux Jr is given by a first-order relation, i.e.
where
k is the reaction rate between Li and Si at the front,
Ci is the Li ion concentration at the interface, and
C0 is the Li concentration in the amorphous phase of Li
3.75Si.
In the steady state, the flux of diffusion at the interface equals that of the interface reaction, viz. J|r=r0 = Jr|r=r0, where r0 is the radius of crystalline silicon. Hence,
| |
 | (10) |
where
y =
R −
r0 is the thickness of Li
xSi.
In an incremental time dt, the number of atoms consumed to form newly lithiated silicon is JrAdt, where A is the cross-sectional area of phase interface. During this time segment, the reaction increases the LixSi layer volume by V0JrAdt, where V0 is the molar volume of Si. As a result, the thickness of lithiated silicon phase increases by dy = V0Jrdt, so that the instantaneous velocity of a phase boundary is given by
| |
 | (11) |
According to assumptions (iii) and (iv), 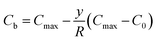 can be obtained. Then, we have
can be obtained. Then, we have
| |
 | (12) |
and
| |
 | (13) |
2.2. Model of lithiation
For simplification of analysis, we here adopt an elastic and perfectly-plastic model to describe the lithiation-induced deformation. The total strain increment, dεij, is taken to be the sum of three contributions, i.e.| | |
dεij = dεcij + dεeij + dεpij,
| (14) |
where subscripts i and j represent directions, and the increment of chemical reaction strain, dεcij, is proportional to the normalized Li concentration, dεcij = βdc, with β the expansion coefficient. The increment of elastic strain, dεeij, obeys Hooke’s law, i.e.| | |
dεeij = [(1 + v)dσij − vdσkkδij]/E,
| (15) |
where E is elastic modulus, v is Poisson’s ratio, δij is the Kronecker symbol with δij = 1 when i = j, and δij = 0 otherwise, and the repeated index means summation.
The increment of plastic strain, dεpij, follows the classic J2-flow rule. The yield function is defined as
| |
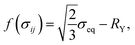 | (16) |
where

is the von Mises equivalent stress with
sij =
σij −
σkkδij/3 the deviatoric stress, and
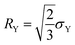
is the radius of the yield surface with the flow stress of
σY. Hence, according to the evolutionary equation,
43 the plastic-strain rate is given by
| |
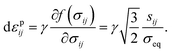 | (17) |
Due to the plastic volume being incompressible, εpii = 0, the consistency parameter can be obtained as
with
| |
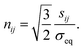 | (19) |
To solve the deformation problem, the material constitutive model should be combined with the force balance
| |
 | (20) |
and strain compatibility condition
| |
 | (21) |
The detailed analysis is as follows: firstly, an initial lithiated thickness is chosen and the Li ion concentration distribution is calculated (see Section 2.1) as a pre-concentration field. Then, with the increase of lithiated thickness, the stress field is calculated based on the step iteration. According to the boundary conditions, the outer surface of Si electrode is traction free, i.e., σr|r=R = 0. Finally, the constitutive equations of elastic-plastic deformation are solved by using a finite element method through the commercial software package (COMSOL v5.0).
3. Results and discussion
3.1. Lithiation process
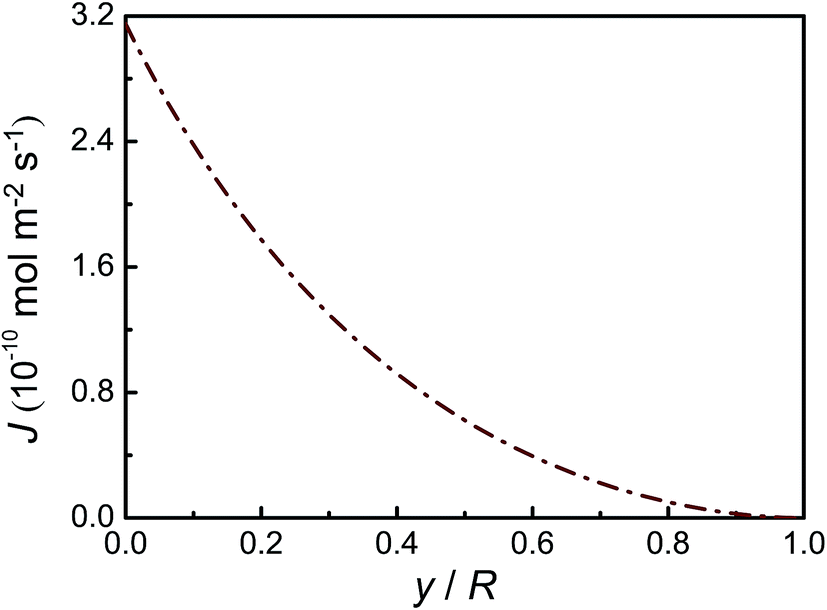
Material parameters used in the model are given in Table 1. As shown in Fig. 2, the surface flux of Li decreases with the lithiated thickness. This phenomenon occurs because the Li ion concentration on surface decreases with migration of Li ions from surface to inside during the charging process. Gradually, the particle will be fully filled with Li ions. Finally, the Li ion flux on the surface drops to zero while the particle is completely lithiated. The concentration distributions of Li on the surface and interface are shown in Fig. 3. Obviously, the surface concentration of Li deceases linearly with lithiated thickness, implying that the LixSi phase changes from Li4.4Si to Li3.75Si due to the Li atom diffusion into the Si anode. The interface concentration of Li is less than the surface concentration of Li, which can provide the driving force for diffusion and interface reaction. Fig. 4 shows the concentration profiles of Li under different lithiated states. It is seen that there is a sharp interface between crystalline Si and amorphous LixSi phases with the core/shell structure, which agrees well with lithiation experiments of Si nanowires and nanoparticles.20
Table 1 Material properties used in the model
| Parameter |
Symbol |
Value |
Reference |
| Diffusivity of Li |
D |
10−16 m2 s−1 |
33 |
| Rate of the diffusion reaction |
k |
2.54 × 10−9 m s−1 |
21 |
| Maximum nominal Li concentration |
Cmax |
0.36 × 106 mol m−3 |
4 |
| Molar volume of Si |
V |
1.20 × 10−5 m3 mol−1 |
21 |
| Young’s modulus of LixSi |
E |
80 GPa |
44 |
| Poisson’s ratio of LixSi |
v |
0.30 |
45 |
| Yield strength of LixSi |
σY |
1 GPa |
34 |
| Expansion coefficient |
β |
0.58 |
4 |
 |
| | Fig. 2 Li ion surface flux as a function of the lithiated thickness during a lithiation process. | |
 |
| | Fig. 3 Li ion concentrations as a function of the lithiated thickness during a lithiation process. | |
 |
| | Fig. 4 The Li ion concentration distributions with various lithiated thicknesses. | |
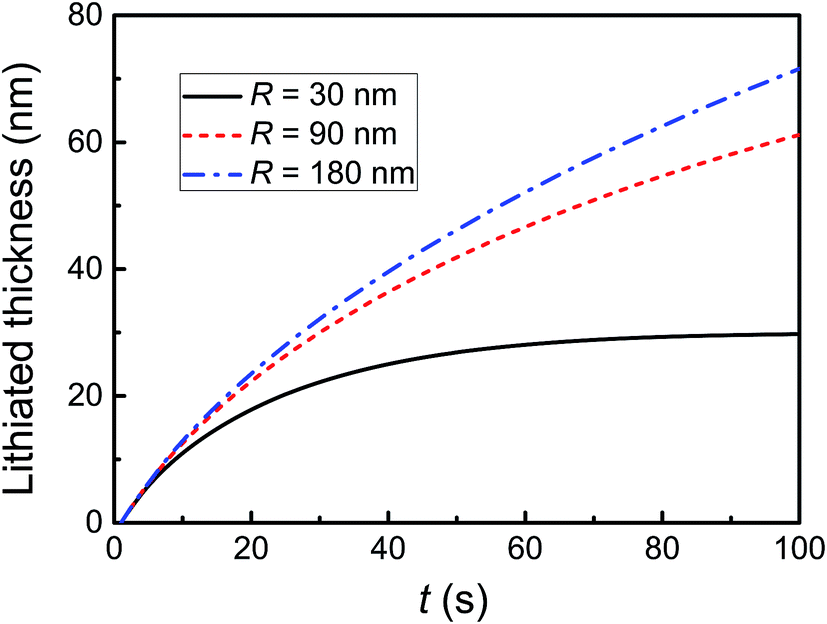
Fig. 5 shows the evolution of lithiated thickness with time. McDowell et al. experimentally observed a similar evolution trend of lithiated thickness.22 It is obvious that the lithiation rate becomes slow no matter how large these particles. When kR/D ≫ 1 and ln(1 − y/R) → −y/R, eqn (12) can be simplified as  . Furthermore, the lithiated thickness is proportional to
. Furthermore, the lithiated thickness is proportional to  , which means lithiation is limited by diffusion. In contrast, when kR/D ≪ 1 and ln(1 − y/R) → −y/R, the lithiated thickness is proportional to t, and lithiation is limited by reaction. Thus, the lithiation rate is related to D and k, as well as the size of materials.
, which means lithiation is limited by diffusion. In contrast, when kR/D ≪ 1 and ln(1 − y/R) → −y/R, the lithiated thickness is proportional to t, and lithiation is limited by reaction. Thus, the lithiation rate is related to D and k, as well as the size of materials.
 |
| | Fig. 5 The evolution of lithiated shell thickness during a lithiation process. | |
Here it is worth noting that, in this kinetic model, the lithiated thickness is based on the initial (rather than deformed) configuration and the diffusion process is assumed as a steady state.
3.2. Stress analysis
When the initial lithiated thickness is fixed, the corresponding concentration and stress distributions can be obtained. As shown in Fig. 6, due to the concentration discontinuity, the hoop stress σθ has a sharp change at interface. As lithiation proceeds, hoop stress becomes tension at a late stage in the surface layer of Si particle. The transition of hoop stress from compression to tension may cause surface cracking of Si electrodes. The radial stress σr on surface is zero based on the traction-free boundary condition. Further, due to symmetry of the system, σr is equal to the hoop stress σθ in the Li-poor phase.
 |
| | Fig. 6 Stress distributions during a lithiation process. | |
4. Conclusions
In this paper, a kinetic model combining the chemical and diffusion reactions has been established to describe the movement of a sharp phase boundary that separates the lithiated and unlithiated materials. The concentration distribution of Li ions, the finial component of lithiated Li3.75Si, and the movement speed of reaction front are quantitatively determined. Furthermore, the stress field is calculated by using an elastic-perfectly plastic model. It is shown that, during the lithiated process, the compressive hoop stress transforms to tension on the lithiated phase surface. The large hoop tensile stress may consequently trigger the morphological instability and fracture in electrodes.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 11372267, 11402086 and 11472141), and the National High Technology Research and Development Program of China (863 Program) (Grant No. 2013AA032502).
References
- A. Mukhopadhyay and B. W. Sheldon, Prog. Mater. Sci., 2014, 63, 58–116 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- K. T. Nam, Science, 2006, 312, 885–888 CrossRef CAS PubMed.
- L. Chen, F. Fan, L. Hong, J. Chen, Y. Z. Ji, S. L. Zhang, T. Zhu and L. Q. Chen, J. Electrochem. Soc., 2014, 161, F3164–F3172 CrossRef CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- P. Lu, C. Li, E. W. Schneider and S. J. Harris, J. Phys. Chem. C, 2014, 118, 896–903 CAS.
- J. Chang, X. Huang, G. Zhou, S. Cui, P. B. Hallac, J. Jiang, P. T. Hurley and J. Chen, Adv. Mater., 2014, 26, 665 CrossRef.
- J. Wang, F. Fan, Y. Liu, K. L. Jungjohann, S. W. Lee, S. X. Mao, X. Liu and T. Zhu, J. Electrochem. Soc., 2014, 161, F3019–F3024 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- L.-F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2008, 9, 491–495 CrossRef PubMed.
- Z. Cui, F. Gao and J. Qu, J. Mech. Phys. Solids, 2012, 60, 1280–1295 CrossRef CAS.
- F. Yang, Mech. Res. Commun., 2013, 51, 72–77 CrossRef.
- J. Zhang, B. Lu, Y. Song and X. Ji, J. Power Sources, 2012, 209, 220–227 CrossRef CAS.
- G. Che, B. B. Lakshmi, E. R. Fisher and C. R. Martin, Nature, 1998, 393, 346–349 CrossRef CAS.
- H. Sun, G. Xin, T. Hu, M. Yu, D. Shao, X. Sun and J. Lian, Nat. Commun., 2014, 5, 4526 CAS.
- M. T. McDowell, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed.
- W. Liang, H. Yang, F. Fan, Y. Liu, X. H. Liu, J. Y. Huang, T. Zhu and S. Zhang, ACS Nano, 2013, 7, 3427–3433 CrossRef CAS PubMed.
- X. H. Liu, J. W. Wang, S. Huang, F. Fan, X. Huang, Y. Liu, S. Krylyuk, J. Yoo, S. A. Dayeh and A. V. Davydov, Nat. Nanotechnol., 2012, 7, 749–756 CrossRef CAS PubMed.
- J. W. Wang, Y. He, F. Fan, X. H. Liu, S. Xia, Y. Liu, C. T. Harris, H. Li, J. Y. Huang and S. X. Mao, Nano Lett., 2013, 13, 709–715 CrossRef CAS PubMed.
- M. J. Chon, V. A. Sethuraman, A. McCormick, V. Srinivasan and P. R. Guduru, Phys. Rev. Lett., 2011, 107, 045503 CrossRef CAS PubMed.
- Z. Cui, F. Gao and J. Qu, J. Mech. Phys. Solids, 2013, 61, 293–310 CrossRef CAS.
- M. T. McDowell, I. Ryu, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed.
- M. Pharr, K. Zhao, X. Wang, Z. Suo and J. J. Vlassak, Nano Lett., 2012, 12, 5039–5047 CrossRef CAS PubMed.
- X. H. Liu, H. Zheng, L. Zhong, S. Huang, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J. H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. Wang, S. X. Mao, Z. Z. Ye, S. Zhang and J. Y. Huang, Nano Lett., 2011, 11, 3312–3318 CrossRef CAS PubMed.
- H. Yang, S. Huang, X. Huang, F. Fan, W. Liang, X. H. Liu, L. Q. Chen, J. Y. Huang, J. Li, T. Zhu and S. Zhang, Nano Lett., 2012, 12, 1953–1958 CrossRef CAS PubMed.
- Y. Lu and Y. Ni, Mech. Mater., 2015, 91, 372–381 CrossRef.
- C. V. Di Leo, E. Rejovitzky and L. Anand, J. Mech. Phys. Solids, 2014, 70, 1–29 CrossRef CAS.
- K. Zhao, M. Pharr, Q. Wan, W. L. Wang, E. Kaxiras, J. J. Vlassak and Z. Suo, J. Electrochem. Soc., 2012, 159, A238–A243 CrossRef CAS.
- K. Zhao, W. L. Wang, J. Gregoire, M. Pharr, Z. Suo, J. J. Vlassak and E. Kaxiras, Nano Lett., 2011, 11, 2962–2967 CrossRef CAS PubMed.
- K. Zhao, G. A. Tritsaris, M. Pharr, W. L. Wang, O. Okeke, Z. Suo, J. J. Vlassak and E. Kaxiras, Nano Lett., 2012, 12, 4397–4403 CrossRef CAS PubMed.
- B. Yang, Y. P. He, J. Irsa, C. A. Lundgren, J. B. Ratchford and Y. P. Zhao, J. Power Sources, 2012, 204, 168–176 CrossRef CAS.
- H. Haftbaradaran and H. Gao, Appl. Phys. Lett., 2012, 100, 121907 CrossRef.
- Z. S. Ma, Z. C. Xie, Y. Wang, P. P. Zhang, Y. Pan, Y. C. Zhou and C. Lu, J. Power Sources, 2015, 290, 114–122 CrossRef CAS.
- K. Zhao, M. Pharr, L. Hartle, J. J. Vlassak and Z. Suo, J. Power Sources, 2012, 218, 6–14 CrossRef CAS.
- K. Zhao, M. Pharr, J. J. Vlassak and Z. Suo, J. Appl. Phys., 2010, 108, 073517 CrossRef.
- Z. Ma, T. Li, Y. Huang, J. Liu, Y. Zhou and D. Xue, RSC Adv., 2013, 3, 7398–7402 RSC.
- I. Laresgoiti, S. Käbitz, M. Ecker and D. U. Sauer, J. Power Sources, 2015, 300, 112–122 CrossRef CAS.
- E. Rejovitzky, C. V. Di Leo and L. Anand, J. Mech. Phys. Solids, 2015, 78, 210–230 CrossRef CAS.
- R. L. Sacci, J. M. Black, N. Balke, N. J. Dudney, K. L. More and R. R. Unocic, Nano Lett., 2015, 15, 2011–2018 CrossRef CAS PubMed.
- G. M. Veith, M. Doucet, J. K. Baldwin, R. L. Sacci, T. M. Fears, Y. Wang and J. F. Browning, J. Phys. Chem. C, 2015, 119, 20339–20349 CAS.
- M. Nie, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 13403–13412 CAS.
- D. E. Arreaga-Salas, A. K. Sra, K. Roodenko, Y. J. Chabal and C. L. Hinkle, J. Phys. Chem. C, 2012, 116, 9072–9077 CAS.
- J. C. Simo and T. J. Hughes, Computational inelasticity, Springer Science & Business Media, 2006 Search PubMed.
- K. Zhao, M. Pharr, S. Cai, J. J. Vlassak and Z. Suo, J. Am. Ceram. Soc., 2011, 94, s226–s235 CrossRef CAS.
- S. Huang, F. Fan, J. Li, S. Zhang and T. Zhu, Acta Mater., 2013, 61, 4354–4364 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 






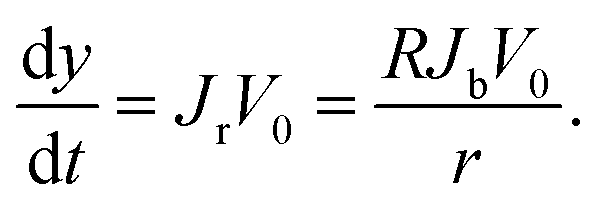
 can be obtained. Then, we have
can be obtained. Then, we have

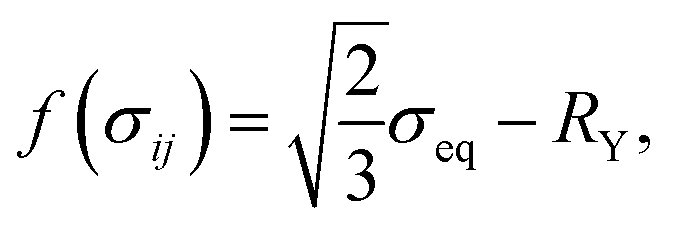
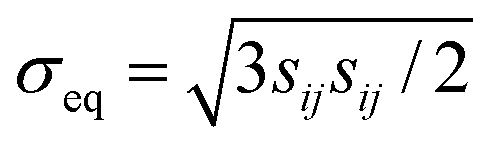 is the von Mises equivalent stress with sij = σij − σkkδij/3 the deviatoric stress, and
is the von Mises equivalent stress with sij = σij − σkkδij/3 the deviatoric stress, and  is the radius of the yield surface with the flow stress of σY. Hence, according to the evolutionary equation,43 the plastic-strain rate is given by
is the radius of the yield surface with the flow stress of σY. Hence, according to the evolutionary equation,43 the plastic-strain rate is given by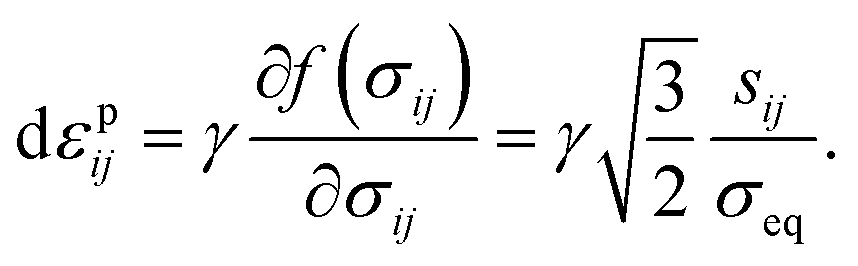
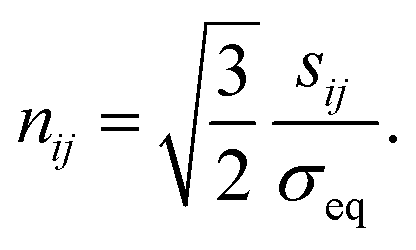




 . Furthermore, the lithiated thickness is proportional to
. Furthermore, the lithiated thickness is proportional to  , which means lithiation is limited by diffusion. In contrast, when kR/D ≪ 1 and ln(1 − y/R) → −y/R, the lithiated thickness is proportional to t, and lithiation is limited by reaction. Thus, the lithiation rate is related to D and k, as well as the size of materials.
, which means lithiation is limited by diffusion. In contrast, when kR/D ≪ 1 and ln(1 − y/R) → −y/R, the lithiated thickness is proportional to t, and lithiation is limited by reaction. Thus, the lithiation rate is related to D and k, as well as the size of materials.