
Catalytic wet air oxidation of high-concentration organic pollutants by upflow packed-bed reactor using a Ru–Ce catalyst derived from a Ru3(CO)12 precursor
Chaoying Yu,
Xu Meng,
Gexin Chen and
Peiqing Zhao*
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: zhaopq@licp.cas.cn; Fax: +86 931 827700
First published on 22nd February 2016
Abstract
Low loading catalysts Ru/γ-Al2O3 and Ru–Ce/γ-Al2O3 (Ru: 0.3 wt%) were prepared by thermolysis of Ru3(CO)12, which exhibited high activity in Catalytic Wet Air Oxidation (CWAO) of high concentration organic pollutants by upflow packed-bed reactor with initial COD 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1. XPS results revealed that two new Ru species (RuA and RuB) were detected due to the chemical interaction between Ru3(CO)12 and the active OH groups on the surface of Al2O3 during the process of Ru3(CO)12 incomplete decomposition, and the reduction of them can lead to more dispersed metallic phases. The TPR spectrum reveals that the catalyst with addition of Ce produced a strong interaction between Ru and the CeO2–Al2O3 support, which was helpful for the catalytic wet air oxidation. Low loading catalyst Ru–Ce/γ-Al2O3 reduced in H2 had the highest activity and the COD removal reached 99.5% above 245 °C. The operating parameters investigated included temperature, reactor pressure, gas flow rate and liquid hourly space velocity (LHSV). The results showed that the COD removal is considerably affected by the temperature; when temperature increased around 2–3 °C, COD removal was improved about 40% due to the obvious exothermic reaction. The experimental results indicated that the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst has excellent activity and stability in the CWAO of high concentration organic pollutants in as packed-bed reactor during 100 hours.
000 mg L−1. XPS results revealed that two new Ru species (RuA and RuB) were detected due to the chemical interaction between Ru3(CO)12 and the active OH groups on the surface of Al2O3 during the process of Ru3(CO)12 incomplete decomposition, and the reduction of them can lead to more dispersed metallic phases. The TPR spectrum reveals that the catalyst with addition of Ce produced a strong interaction between Ru and the CeO2–Al2O3 support, which was helpful for the catalytic wet air oxidation. Low loading catalyst Ru–Ce/γ-Al2O3 reduced in H2 had the highest activity and the COD removal reached 99.5% above 245 °C. The operating parameters investigated included temperature, reactor pressure, gas flow rate and liquid hourly space velocity (LHSV). The results showed that the COD removal is considerably affected by the temperature; when temperature increased around 2–3 °C, COD removal was improved about 40% due to the obvious exothermic reaction. The experimental results indicated that the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst has excellent activity and stability in the CWAO of high concentration organic pollutants in as packed-bed reactor during 100 hours.
Introduction
Wastewater from industries such as pulp and paper, dyeing, chemical, petrochemical, etc. contains a high content of organic matter (chemical oxygen demand (COD = 10–100 g L−1)), or toxic contaminants for which direct biological purification is unfeasible, which can cause severe problems for the environment. They must be treated to satisfy the stringent water quality regulations and the demand for recycling of water in the process. The incapability of conventional methods to effectively remove many organic pollutants has made it evident that new, compact and more efficient systems are needed.Conventional technologies including biological, thermal, and physicochemical treatments have been used to remove the aqueous pollutants.1 Although biological method is widely applied for the treatment of residual wastewaters, it requires a long residence time for microorganisms to degrade pollutants and is not suitable to treat the high concentration organic pollutants as well as the toxic contaminants due to biomass poisoning. Incineration is appropriate for the treatment of effluents having more than 100 g L−1 of chemical oxygen demand (COD). However, it requires extremely high energy and presents considerable emission of other hazardous compounds such as dioxin and furan.2 The aforementioned limitations of conventional methods have encouraged the researchers to develop more efficient and environmental-friendly system for wastewater treatment.
Catalytic wet air oxidation (CWAO) is known to have a great potential for the treatment of effluents containing a high content of organic matter (COD = 10–100 g L−1) or toxic contaminants. CWAO not only reduces the severity of reaction conditions but also more easily decomposes even refractory pollutants, thereby reducing capital and operational cost. CWAO of numerous different single organic compounds has been investigated over the last three decades, using a variety of catalysts, and the CWAO processes were reviewed by many authors.2–7 However, in these studies, most of the initial COD is below 10000 mg L−1. Various solid catalysts including noble metals, metal oxides, and mixed oxides have been widely studied for the CWAO of aqueous pollutants. Generally, catalysts containing noble metals exhibit higher activity than base metal catalyst in CWAO reaction. Supported noble metals (Ru, Pt, etc.) have been developed to reduce the reaction operating conditions of CWAO. These catalysts are potentially active and stable in CWAO of a large range of pollutant effluents containing organic compounds.8–12 For precious metal catalysts, Ru is much cheaper than the other platinum group noble metals, so it has been studied by many scholars. The content of ruthenium in noble metal catalysts usually 1–5 wt% supported on γ-Al2O3,13 CeO2,14 ZrO2,15,16 TiO2,17,18 or carbon.19 Particularly, ruthenium carbonyl used as a catalyst precursor can reduce the Ru loading amount, thus significantly reducing capital costs.20,21
In our previous work, the efficiency of the prepared catalysts was tested in CWAO of different organic compounds with high concentration (COD ≤ 20000 mg L−1) in batch reactor. Accordingly, the main objective of this work was to treat high concentration organic pollutants (initial COD = 200000 mg L−1) in continuous reactor using a Ru catalyst (0.3 wt%) derived from Ru3(CO)12 precursor. The effect of operational conditions such as temperature, reactor pressure, gas flow rate, liquid hourly space velocity (LHSV), and feed concentration as well as catalyst stability were all tested. The metal speciation on the surface of the catalyst and the interaction between metal and carrier was studied and discussed.
Experimental
The catalyst was prepared by incipient wetness impregnation method with hexane solution of Ru3(CO)12 (homemade). γ-Al2O3 was used as a support of the catalysts (BET surface area 180.9 m2 g−1, pore diameter 10 nm, Jiangsu chemical company). Ru–Ce/γ-Al2O3 catalyst was prepared by consecutive impregnation procedure. γ-Al2O3 was added into aqueous solution of Ce(NO3)3·6H2O (AR, Tianjin chemical company), dried at 110 °C for 3 h, and calcined at 500 °C for 6 h, then Ru was supported on the material with the same impregnation, after impregnation for 3 h, the samples were dried for 3 h at 100 °C, and then introduced in a tubular quartz reactor and reduced under flowing hydrogen (20 L h−1) at 400 °C for 6 h. The content of Ru and Ce in all catalysts was fixed at 0.3 and 3 wt% relative to the weight of γ-Al2O3, respectively.X-ray photoelectron spectroscope (XPS) was measured with VG 82 ESCALAB 210 instrument, employing Mg Kα radiation. All the binding energies were calibrated by using Al 2p (74.5 eV) as a reference, and spectral peaks were fitted using a mixed Gaussian–Lorentzian line shape and Shirley baselines. More detailed characterization procedures can be found in previous work.22
TPR experiments were carried out with the aid of a temperature programmed ChemiSorb PCA-1200 instrument (Builder, China) equipped with a TCD detector. Prior to TPR measurements, the sample (about 200 mg) was treated in situ as follows: under flowing oxygen the temperature was raised at 20 K min−1 to the desired value and maintained at that level for an additional 10 min after which it was cooled to room temperature in flowing N2. The TPR experiments were performed with a mixture of 5% H2 in Ar. The flow rate of the reducing gas was 30 mL min−1. The temperature increased from room temperature to 573 K at a linearly programmed rate of 5 K min−1, and then the temperature was raised to 1123 K at rate of 10 K min−1.
Experiments were carried out in a micro pilot packed-bed reactor schematically described in Fig. 1. Air from a gas cylinder was depressurized to 4 MPa and its flow rate adjusted with a mass flow controller (Sevenstar D07) to give a constant flow of 100–500 mL min−1. Aqueous solutions of organic pollutants were fed with a high-pressure pump (SSI LabAlliance Series II Pump) (0.01–10 mL min−1). Air and liquid were mixed at the inlet of the reactor which was equipped with an axially located thermocouple and heated with an electrical furnace (length, 430 mm; internal diameter, 12 mm; internal volume, 48 cm3). The reactor was packed with the catalyst placed between two layers of metal-free, supporting material. Gas and liquid were flown in cocurrent upflow mode through the reactor. At the reactor outlet the liquid and gas flows were cooled and recovered in the gas–liquid separator. The gas flow was depressurized to atmospheric pressure by a backpressure regulator (Swagelook KBP1N0A400P20000). The reactor loaded with 10 mL of 0.3% Ru–Ce/γ-Al2O3 catalyst was operated under the following conditions. It was pressurized with 3 MPa of air or oxygen flowing at 100–300 mL min−1. When the set temperature was attained the aqueous solution of high concentration organic pollutants (initial COD = 200000 mg L−1) was pumped at flow rate (30 mL h−1) to saturate the catalyst with the liquid and to fill partially the gas–liquid separator, and then the liquid flow rate was reduced to standard operating conditions (15 mL h−1) and the regulation of liquid level in the separator was activated. Samples were periodically withdrawn from the reactor to measure the COD with a 5B-3B COD analyzer. In all cases, the error of COD analysis was never greater than ±2.0%.
 | ||
| Fig. 1 Packed-bed reactor setup. | ||
The catalyst life evaluation test of 100 h was carried out in a packed-bed reactor with upflow of high concentration pollutants (initial COD = 200000 mg L−1) and O2. The reaction condition was as follows: reaction temperature = 245 °C, reaction pressure = 3.0 MPa, LHSV = 1.5 h−1, O2 flow rate = 150 mL min−1.
Activities of the CWAO catalysts were tested using model compounds (Alpha) mixture of isopropyl alcohol, methanol and phenol (one of the most toxic pollutants).
Results and discussion
XPS was used to study the surface coverage of the ruthenium species on the catalysts. Fig. 2 shows a typical XPS spectrum for the 0.3% Ru catalysts reduced in H2 and calcined at 673 K. Fig. 2a is the XPS 3d spectrum of the 0.3% Ru/γ-Al2O3 reduced at 673 K in H2. The Ru 3d core level shows the presence of Ru0 species at 280.1 eV and RuO2 at 281.7 eV, and the amount of RuO2 is larger than that of Ru0. It is observed that the sample was completely decomposed at 673 K. The small peak at 288.1 eV probably attributed to the carbonaceous deposits on the surface of the sample.23 | ||
| Fig. 2 XPS for the Ru 3d spectra for (a) Ru3(CO)12 on γ-Al2O3 reduced at 673 K in H2. (b) Ru3(CO)12 on γ-Al2O3 calcined at 673 K. (c) Ru3(CO)12 on 3% Ce/γ-Al2O3 reduced at 673 K in H2. | ||
Fig. 2b shows the Ru 3d spectrum for the Ru3(CO)12 on γ-Al2O3 calcined at 673 K. The Ru 3d5/2 peaks at 281.1 eV belongs to RuO2 and 282.7 eV might be attributed to a new subcarbonylic species RuB due to the carbonyl ruthenium incomplete decomposition on the support. Fig. 2c shows the peak at 283.1 eV in 0.3% Ru/3% Ce/γ-Al2O3 perhaps belongs to another new subcarbonylic species RuA. It was proved that two new Ru species (RuA and RuB) were detected during the Ru3(CO)12 decomposition process due to chemical interaction with the active OH groups on the surface of Al2O3 support, and the reduction of them can lead to more dispersed metallic phases.24 Detailed results for the catalyst characterization can be consulted in previous publications.22
H2-TPR is conducted over four catalysts to understand the reduction behaviors of Ru oxides in the reduced and calcined catalysts, as shown in Fig. 3. With regard to 0.3% Ru catalyst without adding Ce, the presence of two peaks in the TPR spectrum following oxidation at different temperature (see Fig. 3a and b) suggests the existence of a bimodal distribution of particle sizes. The main reduction peak for Ru catalyst presents at 372 K and 403 K. The reduction of a relatively small number of large RuOx particles with a metallic core gives rise to the peak at lower temperature (372 K). The high temperature peak at about 403 K is the result of the reduction of the completely oxidized smaller RuOx or RuO2 particles.25,26
 | ||
| Fig. 3 TPR profiles of the catalysts after oxidized at different temperature (Ru: 0.3 wt%). (a) Ru/γ-Al2O3, reduced in H2. (b) Ru/γ-Al2O3 calcined. (c) Ru/3% Ce/γ-Al2O3 reduced in H2 (d) Ru/3% Ce/γ-Al2O3 calcined. | ||
As for the samples oxidized at 573 K in Fig. 3a and b, the results indicate no further segregation of the large RuOx particles and small oxide particles takes place. The larger peak at 372 K after oxidation above 673 K indicate that this treatment results in a lot of large RuOx or RuO2 particles. The main reduction peak for Ru catalyst presents at 372 K after oxidation above 973 K is the result of the reduction of the completely oxidized smaller particles.
However, the 0.3% Ru/3% Ce/γ-Al2O3 catalysts have shown three reduction peaks in Fig. 3c and d, the third high temperature peak at 430–460 K due to ruthenium strongly interacted with CeO2 and Al2O3. The maximum of the third peak shifts to higher temperatures with increasing oxidation temperature. The oxidation at higher temperatures to promote segregation of oxide phases. The calcined samples after oxidation 573 K and 673 K show two wide reduction peaks (600–900 K) that are connected with the surface oxygen.27
As mentioned above, the catalysts with adding Ce have a strong interaction between Ru and Ce–Al. This strong interaction is more obvious in the low load catalyst in Fig. 4a, the third peak intensity is higher after oxidation above 773 K. But the third peak almost did not appear in 0.5 wt% Ru catalyst after oxidation at 773 K in Fig. 4b, it appears oxidized above 873 K, low intensity may be due to the surface of the catalyst dispersed Ru covering. The peak at 403 K higher than at 372 K indicates the small RuOx/RuO2 is majority in 0.5 wt% Ru catalyst after oxidation at 773 K and 873 K.
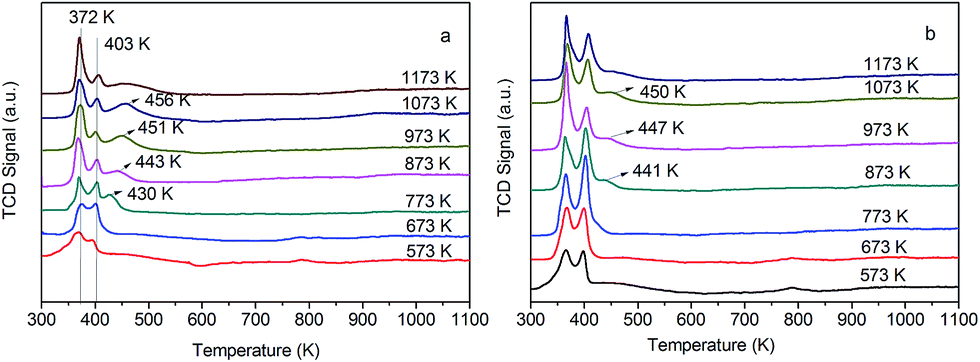 | ||
| Fig. 4 TPR profiles of the 0.3 wt% (a) and 0.5 wt% (b) Ru catalysts after oxidized at different temperature. | ||
The prepared Ru/γ-Al2O3 and Ru–Ce/γ-Al2O3 catalysts using the precursor Ru3(CO)12 were tested in the CWAO of highly concentrated organic compounds mixture of isopropyl alcohol, methanol and phenol (initial COD = 200000 mg L−1). Fig. 5 shows that the COD removal is only about 20% in blank test over Ce/γ-Al2O3 catalyst without Ru loading. The activity of reduced catalyst is slightly higher than that of the calcined catalyst, possibly due to the reduction of the dispersion of the metal after calcined. The COD removal were about 95% on calcined 0.3% Ru–Ce catalyst and 98% on 0.3% Ru–Ce reduced in H2. Based on XPS analysis, it comes to the conclusion that the addition of Ce can greatly increase the activity of the catalyst due to new subcarbonylic species RuA on the surface of Al2O3 support and reduction of them can lead to more dispersed metallic. The COD removal on Ru–Ce/γ-Al2O3 was about 10–15% higher than that on Ru/γ-Al2O3 due to the addition of Ce not only improved the dispersion of the active component, but also produced a strong interaction with the CeO2–Al2O3 support as shown in H2-TPR characterization, which was helpful for the catalytic wet air oxidation.
 | ||
| Fig. 5 Ru and Ru–Ce catalytic activity test. | ||
Temperature is also a very important parameter in the CWAO of organic compounds. Firstly, according to Arrhenius' law28 the higher the reaction temperature is, the faster the reaction is. Secondly, above 100 °C, the oxygen solubility in water increases with temperature. For the treatment of high concentration organic wastewater, a slight change could bring about an impact on the reaction results because of obvious exothermic phenomenon during the reaction process. Some organic matter in the oxidation reaction activation energy is lower the faster the reaction speed, exothermal also evident, for provides the required heat for the reaction, also possibly mixes the organic matter to have the cooxidation function in the oxidation process. Results in Fig. 6a shows that COD removal was very sensitive to the temperature over 0.3 wt% Ru/3% Ce/γ-Al2O3. The COD removal was only about 47% at the beginning of the reaction at 240 °C (3.0 MPa, 1.5 h−1, O2 = 150 mL min−1), and then, the COD removal enhanced slowly along with the reaction time and reached 92.6% after 360 min. However, when the temperature improved to 243 °C, the COD removal can reached 98% and 99.5% above 245 °C. Taking into consideration the abovementioned multisided reasons, we chose 245 °C as the optimum temperature for the study of the CWAO of high concentration organic pollutants over the 0.3% Ru–Ce/γ-Al2O3 catalyst.
 | ||
| Fig. 6 Influence of the reaction conditions on COD removal ((a) reaction temperature. (b) Pressure. (c) O2 flow rate. (d) LHSV). | ||
Fig. 6b shows that the effects of increasing gas pressure on COD removal under 245 °C, 1.5 h−1 (LSHV) and 150 mL min−1 (O2 flow rate). A significant improvement in COD removal occurred when pressure changed from 2.0 to 3.0 MPa, the COD removal improved from 91% to 98%. When pressure changed from 3.0 to 4.0 MPa, the change of the COD removal was very small and improvement was only 2%. This finding implies that the effect of pressure diminishes when pressure exceeded 3.0 MPa, because the reaction becomes liquid reactant limited at high pressure. Further increase in the reactor pressure, and hence liquid phase oxygen concentration, will have a minimal effect since oxygen is not the limiting reactant anymore. Elevated levels of pressure and temperature improve the solubility of oxygen. This facilitates the rate of mass transport to the wetted catalyst surface. The gas–liquid interfacial area and the liquid-side volumetric mass-transfer coefficients, depends on pressure (gas density).29 Hence, due to the enhanced gas–liquid interaction and interphase mass transfer, an increased gas pressure is favourable for the performance of upflow reactors.
Fig. 6c shows that the COD removal was enhanced by increasing the oxygen velocity at high reactor pressure. The COD removal was found to increase almost linearly with an increase in the oxygen velocity under 245 °C, 3.0 MPa, LHSV 1.5 h−1. When the oxygen flow rate was 50 mL min−1, COD removal of less than 60%, and when the oxygen flow rate increased to 100 mL min−1 or more, COD removal can reach 98%, but the initial reaction is slow. The strong dependence was due to a linear increase in the gas–liquid and liquid–solid mass transfer coefficients with the gas velocity.30 However, the effect of gas velocity on reactor performance diminished when gas velocities became moderate. So 150 mL min−1 is the most appropriate oxygen flow.
As previously commented, a decrease in the liquid flow rate resulted in positive effects on COD removal. Fig. 6d shows the COD removal decreased with an increase in LHSV under 245 °C, 3.0 MPa, O2 flow rate 150 mL min−1 attaining COD removal of 98.2% at LHSV = 1.5 h−1. Then, changing the LHSV to 3.0 h−1, the COD removal decreased to around 92%. A lower feed flow rate is preferred because the longer residence time will benefit for the completing degradation of organic matter. Although a high liquid superficial velocity enhances the mass transfer coefficient, it actually lowers the liquid reactant residence time, which corresponds to the decreased COD removal. For gas-limited reactions, scale-up at constant LHSV can lead to very poor performance as the catalyst effectiveness factor drops with increased contacting efficiency due to a reduction in the gas reactant supply.
Fig. 7 reveals the activity for CWAO of high concentration organic pollutants over the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst. The catalyst displays higher activity in the CWAO of high concentration organic pollutants (initial COD = 200000 mg L−1, 245 °C, 3.0 MPa, 1.5 h−1, O2 = 150 mL min−1), and 98–99% COD was obtained during 20 h reaction, then decreased slowly and stabilized around 92% to 96%. These experiments show that despite the formation of carbonaceous compounds onto the catalyst surface, it presents a good stability up to 210 h in the considered reaction conditions. From the experimental results in the packed-bed reactor, it indicated that the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst possesses excellent activity and stability in the CWAO of high concentration organic pollutants.
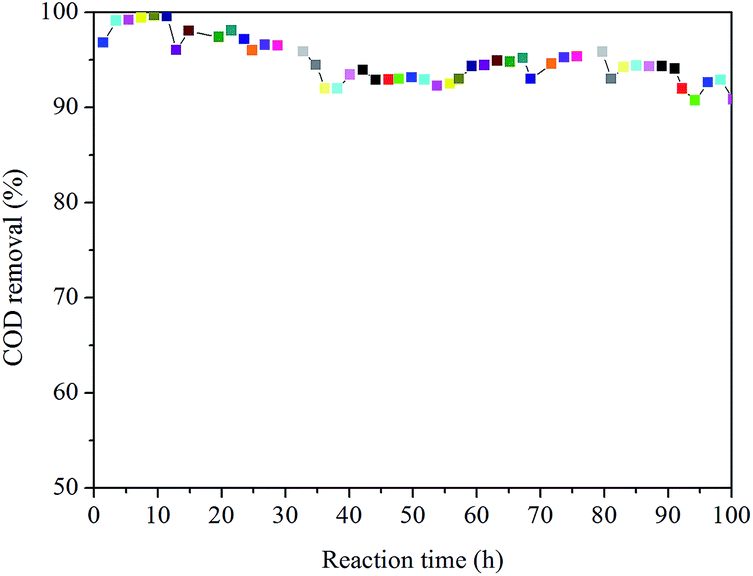 | ||
| Fig. 7 Stability of the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst in the CWAO of high concentration organic pollutants during 100 hours. | ||
Since the catalyst activity is high, the degradation of organic matter quickly, rarely generates intermediate, which decreases the deposition of carbonaceous compounds to some extent and maintains the stability of the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst in the CWAO of high concentration organic pollutants.
Conclusions
XPS results revealed that two new Ru species (RuA and RuB) were detected due to the Ru3(CO)12 chemical interaction with the active OH groups on the surface of Al2O3 support during the Ru3(CO)12 incomplete decomposition process, reduction of them can lead to more dispersed metallic. The catalyst with addition of CeO2 not only improved the dispersion of the active component, but also produced a strong interaction with the CeO2–Al2O3 support as shown in H2-TPR characterization, which was helpful for the catalytic wet air oxidation.The low loading Ru–Ce (Ru: 0.3 wt%) catalyst reduced in H2 had the highest activity in the CWAO of high concentration organic pollutants (initial COD = 20000 mg L−1). In high concentration organic matter oxidation process, since the intense exothermic reaction, slight changes in temperature have great effects on the reaction. When the temperature improved from 240 °C to 243 °C, the COD removal increased from 47% to 98% and can reached 99.5% above 245 °C at 3.0 MPa, 150 mL min−1 (oxygen flow rate) and LHSV of 1.5 h−1.
The experimental results indicated that the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst is the excellent activity and stability in the CWAO of high concentration organic pollutants (initial COD = 200000 mg L−1) in the packed-bed reactor during 100 hours. Since the catalyst activity is high, the degradation of organic matter quickly, rarely generates intermediate, which decreases the deposition of carbonaceous compounds to some extent and maintains the stability of the 0.3 wt% Ru–Ce/γ-Al2O3 catalyst in the CWAO of high concentration organic pollutants.
Acknowledgements
The authors gratefully acknowledge the financial support of the Natural Science Foundation of China, No. 21307139.Notes and references
- I. Oller, S. Malato and J. A. Sánchez-Pérez, Sci. Total Environ., 2011, 409, 4141–4166 CrossRef CAS PubMed.
- K. H. Kim and S. K. Ihm, J. Hazard. Mater., 2011, 186, 16–34 CrossRef CAS PubMed.
- F. Luck, Catal. Today, 1996, 27, 195–202 CrossRef CAS.
- S. Imamura, Ind. Eng. Chem. Res., 1999, 38, 1743–1753 CrossRef CAS.
- J. T. Suresh, K. Bhargava, J. Prasad, K. Föger, D. B. Akolekar and S. C. Grocott, Ind. Eng. Chem. Res., 2006, 45, 1221–1258 CrossRef.
- A. Cybulski, Ind. Eng. Chem. Res., 2007, 46, 4007–4033 CrossRef CAS.
- J. Levec and A. Pintar, Catal. Today, 2007, 124, 172–184 CrossRef CAS.
- J. Gaalova, J. Barbier Jr and S. Rossignol, J. Hazard. Mater., 2010, 181, 633–639 CrossRef CAS PubMed.
- C. D. Taboada, J. Batista, A. Pintar and J. Levec, Appl. Catal., B, 2009, 89, 375–382 CrossRef CAS.
- M. Triki, Z. Ksibi, A. Ghorbel and F. Medina, Microporous Mesoporous Mater., 2009, 117, 431–435 CrossRef CAS.
- D. Phamminh, P. Gallezot and M. Besson, Appl. Catal., B, 2007, 75, 71–77 CrossRef.
- A. Song and G. Lu, RSC Adv., 2014, 4, 15325 RSC.
- S. Yang, Y. Feng, J. Wan, W. Zhu and Z. Jiang, Appl. Surf. Sci., 2005, 246, 222–228 CrossRef CAS.
- S. Roy and A. K. Saroha, RSC Adv., 2014, 4, 56838–56847 RSC.
- G. Sun, A. Xu, Y. He, M. Yang, H. Du and C. Sun, J. Hazard. Mater., 2008, 156, 335–341 CrossRef CAS PubMed.
- H. Wei, X. Yan, S. He and C. Sun, Catal. Today, 2013, 201, 49–56 CrossRef CAS.
- M. Bistan, T. Tisler and A. Pintar, Catal. Commun., 2012, 22, 74–78 CrossRef CAS.
- S. Yang, W. Zhu and X. Wang, Rare Met., 2011, 30, 488–495 CrossRef CAS.
- P. Gallezot, S. Chaumet, A. Perrard and P. Isnard, J. Catal., 1997, 168, 104–109 CrossRef CAS.
- S. D. A. Beck and L. Guczi, Inorg. Chem., 1998, 27, 3220–3226 CrossRef.
- Y. I. Yasuo Izumi and K.-ichi Aika, J. Phys. Chem., 1996, 100, 9421–9428 CrossRef.
- C. Yu, P. Zhao, G. Chen and B. Hu, Appl. Surf. Sci., 2011, 257, 7727–7731 CrossRef CAS.
- J. Wang, W. Zhu, X. He and S. Yang, Catal. Commun., 2008, 9, 2163–2167 CrossRef CAS.
- A. Z. A. E. Gugelielminotti, J. Catal., 1982, 74, 225–239 CrossRef.
- J. M. Rynkowski, T. Paryjczak and M. Lenik, Appl. Catal., A, 1995, 126, 257–271 CrossRef CAS.
- D. E. Damiani, E. D. Perez Millan and A. J. Rouco, J. Catal., 1986, 101, 162–168 CrossRef CAS.
- J. M. Rynkowski, T. Paryjczak, A. Lewicki, M. I. Szynkowska, T. P. Maniecki and W. K. Jozwiak, React. Kinet. Catal. Lett., 2000, 71, 55–64 CrossRef CAS.
- W.-M. Liu, Y.-Q. Hu and S.-T. Tu, J. Hazard. Mater., 2010, 179, 545–551 CrossRef CAS PubMed.
- M. H. Al-Dahhan, F. Larachi, M. P. Dudukovic and A. Laurent, Ind. Eng. Chem. Res., 1997, 36, 3292–3314 CrossRef CAS.
- F. Stuber, A. M. Wilhelm and H. Delmas, Chem. Eng. Sci., 1996, 51, 2161–2167 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |