
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiolate-protected golden fullerenes. A 32-ve core involving a hollow Au32 cage†
M. Rauhalahtia,
A. Muñoz-Castro*b and
D. Sundholm*a
aDepartment of Chemistry, University of Helsinki, P. O. Box 55 (A. I. Virtanens Plats 1), FIN-00014, Finland. E-mail: sundholm@chem.helsinki.fi
bGrupo de Química Inorgánica y Materiales Moleculares, Universidad Autonoma de Chile, El Llano Subercaseaux 2801, Santiago, Chile. E-mail: alvaro.munoz@uautonoma.cl
First published on 17th February 2016
Abstract
We have computationally investigated the possible formation of large hollow gold nanostructures based on a Au32 core covered with a thiolate layer using relativistic density functional theory calculations. We have found that [Au32@Au12(SR)18]6− is a plausible candidate that retains the structural, electronic and spherical aromatic properties of the Au32 cage of its parent bare Au32 golden fullerene. The study shows that the low-energy part of the optical spectrum can serve as a guide to identify such hollow gold structures among other small-sized gold nanoclusters with similar nuclearities. The low-lying excited states are dominated by 1f → 1g transitions, which significantly distinguish hollow clusters from other nanoclusters, like the prominent [Au25(SR)18]− cluster, whose low-lying excitations are dominated by 1p → 1d transitions. The gold nanoclusters studied here can serve as model compounds for assessing the metallic core size effects on the absorption energies and the influence of the surface structure of the gold core on the cluster properties. The present study suggests that it is also plausible that even larger hollow structures derived from Au42, Au72, and Au92 golden fullerenes can exist.
Introduction
Besides their aesthetic contextures, gold nanostructures have also attracted great interest as fundamental building blocks in nanoscience, where novel metal clusters with variable sizes play important roles for the development of technological applications such as nanoelectronics, nanosensors, and for biomedicine.1–4 Computational studies of the molecular and electronic structures of stable gold clusters have contributed to rationalizations of their optical, chiroptical, and catalytic properties5–10 and provided a detailed understanding of the molecular properties of novel clusters with extraordinary high stability. Such studies on thiolate-protected gold nanoclusters have led to characterizations of a number of atomically identified nanoclusters with strong gold–gold and gold–sulphur interactions, which yield cluster structures that are stable against degradation.1,4,11 The thiolate substituted gold clusters have interesting electronic structures whose properties originate from the inner metallic core embedded in the protecting layer composed of several stabilizing thiolate groups. The stability of the overall cluster originates from the electronic and structural states of the metallic core with a magic number of valence electrons (ve) as previously shown for Aun(SR)m clusters such as Au25(SR)18, Au36(SR)24, Au38(SR)24, and Au144(SR)60 (ref. 8 and 12–16) (Fig. 1). [Au25(SR)18]− and [Au144(SR)60] clusters have nearly spherical cores with very small distortions from the ideal icosahedral structure.8,16 They display bulk-like fcc structures,15 which is common for many gold clusters with core masses between 4.9 and 28.3 kDa. The Au144(SR)60 cluster has several concentric structural layers as seen from its Au12 + Au42 + Au60 contexture that is evaluated theoretically.16 X-ray experiments showed that Au133(SR)52 with a Au + Au12 + Au42 + Au52 core has a similar layer structure.17 The chemistry of gold compounds and gold clusters is strongly influenced by relativistic effects18 leading to unusual gold cluster structures such as tetrahedral Au20 (ref. 19) and the proposed golden fullerene consisting of an Au32 cage cluster.20 Recently, the thermal stability of the hollow icosahedrons structure of Au32 has been studied computationally suggesting that such conformation is the ground state up to 300 K, remaining in a hollow conformation up to 400 K.21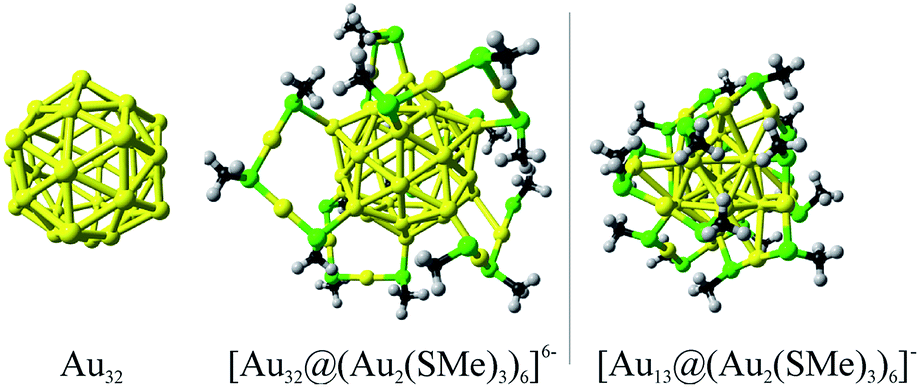 | ||
| Fig. 1 Schematic representation of the thiolate protected version of the golden fullerene Au32, in comparison to [Au25(SMe)18]− cluster, denoting its core based structure. | ||
To understand how the electronic states of relatively small nanoclusters depend on the cluster structure, a variety of gold cluster structures with high symmetry has to be investigated. Constructions of Au32 based substituted gold clusters enable systematic studies of the molecular structures and how they affect other molecular properties of hollow gold nanoclusters. In this work, we elucidate whether a thiolate-protected hollow gold Au32 nanostructure bearing 32 valence electrons can be made, which has not been experimentally characterized to date. We also investigate whether formation of larger-sized hollow cluster structures with more valence electrons in the inner metallic core is feasible.
Computational details
Structural optimizations and subsequent property calculations were performed at the relativistic density functional theory (DFT) level using the ADF 2014 program package.22 The all-electron triple-ξ Slater basis set plus the double-polarization (STO-TZ2P) basis functions were used in combination with the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) functional.23 The use of the PBE functional renders direct comparisons with other computational studies of gold nanoclusters feasible.16,23–26 Relativistic effects were taken into account at the ZORA level (PBE-ZORA/TZ2P).27,28 Environmental effects were considered by using the conductor-like screening model29 (COSMO) with a dielectric constant ε = 13.5 and an atomic gold radius of 3.5 Å. Such parameters correspond to dimethylformamide, an aprotic polar solvent employed able to solvate high charged clusters. The electronic excitation energies were calculated at the PBE level as in previous similar studies.25,26 The optical spectra were also calculated at the DFT level using the asymptotically corrected Leeuwen–Baerends (LB94) functional.30 A comparison of the LB94 and PBE level calculations for [Au25(SMe)18]− in the ESI† section shows that similar results are obtained with the two functionals.Results and discussion
The molecular structure of the thiolate-protected Au32 cluster (Fig. 1) was evaluated according to the “divide and protect” concept, which is a powerful approach to predict conformations of small and medium sized nanostructures.6 The resulting protected cluster have similar structural moieties as obtained for [Au25(SR)18]− (R = methyl) with six dimeric RS–Au–RS–Au–RS (Au2(SR)3) units attached to each vertex of the icosahedral cage. For the Au32 core, the most favorable conformation is obtained by protecting the twelve vertices, which are reachable by six Au2(SR)3 units resulting in an overall Au44(SMe)18 cluster. According to the electron count rules given by Häkkinen,6 the overall cluster reach a closed-shell configuration in its −6 form, resulting in a [Au32@Au12(SMe)18]6− cluster with a 32-ve core (n* = Nva − M − z = 44 − 18 + 6 = 32 ve).6The structure of the bare Au32 cluster consists of concentric icosahedron and dodecahedron cages with radii of 4.513 and 3.984 Å, respectively (Fig. S1†). In the thiolate-protected counterpart, the inclusion of Au2(SR)3 units leads to a small increase in the average icosahedral-section radius of 0.206 Å, whereas the radius in the complementary dodecahedron section decreases on the average by about −0.124 Å. Thus, the golden fullerene structure is retained in the [Au32@Au12(SMe)18]6− cluster, which is a structure that can be obtained in synthesis using polar solvents owing to its −6 negative charge,31 similar to the synthesis of the [Ag32@Ag12(SR)30]4− (ref. 32) clusters.
In comparison with the [Au25(SMe)18]− (ref. 8) and [Au144(SMe)60] (ref. 16) clusters having an endohedral and a hollow icosahedron Au12 cage with radii of 2.854 and 2.706 Å, respectively, the [Au32@Au12(SMe)18]6− structure has the possibility to form a hollow cage involving a larger deltahedron core with an average inner radius of 4.201 Å. Hence, a large inner diameter of about 0.9 nm is obtained for the thiolate-protected counterpart of the Au32 golden fullerene20 as compared to a radius of 0.5 nm for the Au12 cage. The proposed cluster structure exhibits a core that differs from the usual bulk-like fcc structures found in many thiolate-protected clusters.
The radial distribution of gold atoms for these structures is shown in Fig. 2. The figure shows that the size of the icosahedral section of the thiolate-protected Au32 cluster is able to allocate the first and second gold layers of [Au144(SMe)60] yielding a Au12 + Au42 structure that represents about 50% of the gold atoms of its Au114 core. The gold atoms at the protecting layer for the Au32 based cluster are located about 7.3–7.9 Å from the center of the structure, which is indeed in the range of the observed one of 9.0 Å for [Au144(SMe)60], suggesting that one might obtain large gold clusters with fewer gold atoms than expected for a compact cluster. The core mass of the gold atoms in the [Au32@Au12(SMe)18]6− cluster is 8.7 kDa showing that is possible to construct structural features of heavier gold clusters from lighter ones. Such a core can be considered as an inverted case of the 18-ve excavated-dodecahedron architecture found in [Ag32@Ag12(SR)30]4−,32 and the first structural shell of [Ag92@Ag60(SR)60]2+.33 Moreover, methyl groups can be replaced by larger protecting group in order to cover the entire core shell.
 | ||
| Fig. 2 Radial distribution of gold atoms in thiolate protected version Au32, in comparison to [Au25(SMe)18]− and [Au144(SMe)60], denoting its core based structure. | ||
According to the super atom approach,5–7 the 32-ve in the bare Au32 cluster has a 1s21p61d101f14 electronic configuration, composed solely by a first set of super atomic shells due to the single structural layer.34 The electron configuration can be understood by considering that the radial part of the effective potential of the shell is very narrow implying that orbitals with radial nodes are much higher in energy than the next orbital shell with larger angular momentum quantum number and additional angular nodes. For example, the 1d shell is therefore energetically below 2p. Isosurfaces of relevant levels are depicted in Fig. 3, representing shells with increasing number of angular nodes. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have 1f and 1g characters, respectively. The 1f and 1g shells are split due to symmetry reasons. The icosahedral point group (Ih) has degenerate levels up to five-fold shells. The 1f block is therefore composed into one four-fold and one three-fold degenerate levels. The nine orbitals of the 1g shell are analogously split into two blocks consisting of four-fold and a five-fold degenerate orbitals.
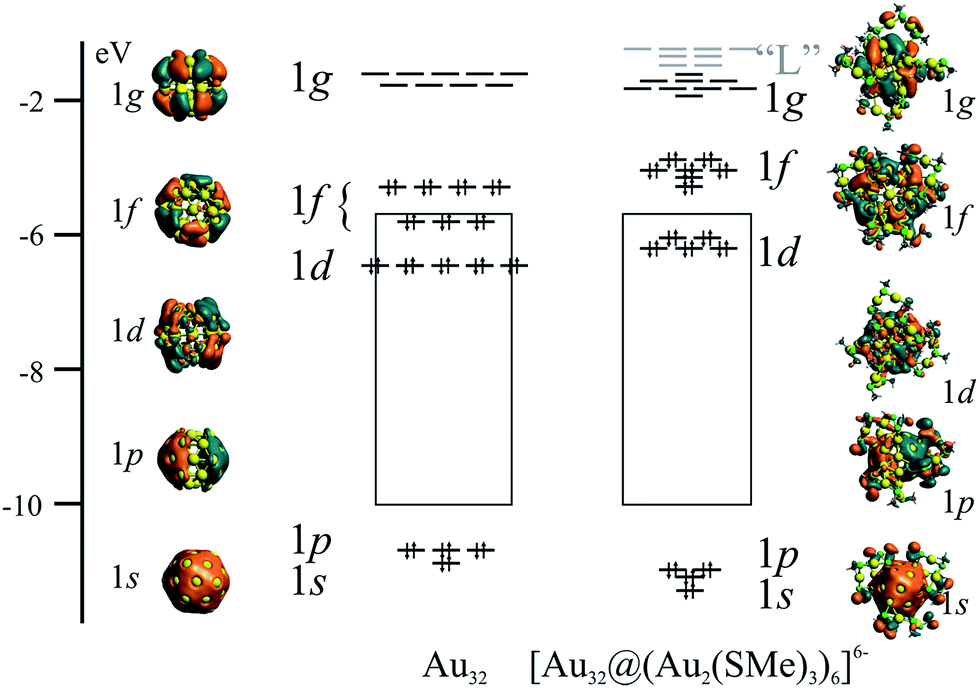 | ||
| Fig. 3 Super-atomic shells of Au32, denoting their counterparts in the [Au32@(Au2(SMe)2)6]6− cluster. | ||
The addition of the protecting groups does not significantly affect the superatomic shell structure. However, some of the orbital contribution originate from the protecting layer. The resulting 1p and 1d shells have lower symmetry, as shown by the reduced degeneracy (Fig. 3). The frontier orbitals form a block of seven high-lying occupied orbitals of 1f character, whereas the low-lying unoccupied orbitals have 1g character. The higher unoccupied orbitals are mainly located to the protecting layer. They are denoted “L” in Fig. 3 for clarity. Thus, the frontier orbitals and the electronic structure of the protected cluster retain the main features of the bare Au32 cluster. This observation suggests the possibility to extend the here described superatomic shell structure to the endohedrally-doped golden fullerenes counterparts,35 allowing the balance of the overall negative charge from endohedral atoms.
Optical properties of thiolate-protecting gold clusters have previously been investigated.8,11,25,26 Computational studies of optical properties are useful for characterizing distinctive patterns for different gold clusters, which can be compared with measured absorption spectra when experimental data are available. To evaluate the characteristic fingerprint of the [Au32@Au12(SMe)18]6−, cluster, the low-energy part (up to 2.0 eV) of the electronic absorption spectrum of [Au32@Au12(SMe)18]6− was calculated and compared to the corresponding spectra of [Au25(SMe)18]−, denoted as [Au13@Au12(SR)18]− hereafter (Fig. 4). The nature of the frontier orbitals for the clusters leads to different selection rules for the low-energy absorption spectra of the studies clusters. The low-energy transitions of [Au32@Au12(SMe)18]6− are mainly dominated by 1f → 1g transitions, whereas for [Au13@Au12(SR)18]− the corresponding transitions are dominated by the 1p → 1d character, which has previously been discussed by Jiang et al.24 Thus, the hollow gold clusters discussed in this work provide a novel extension of optical properties of gold nanoparticles.
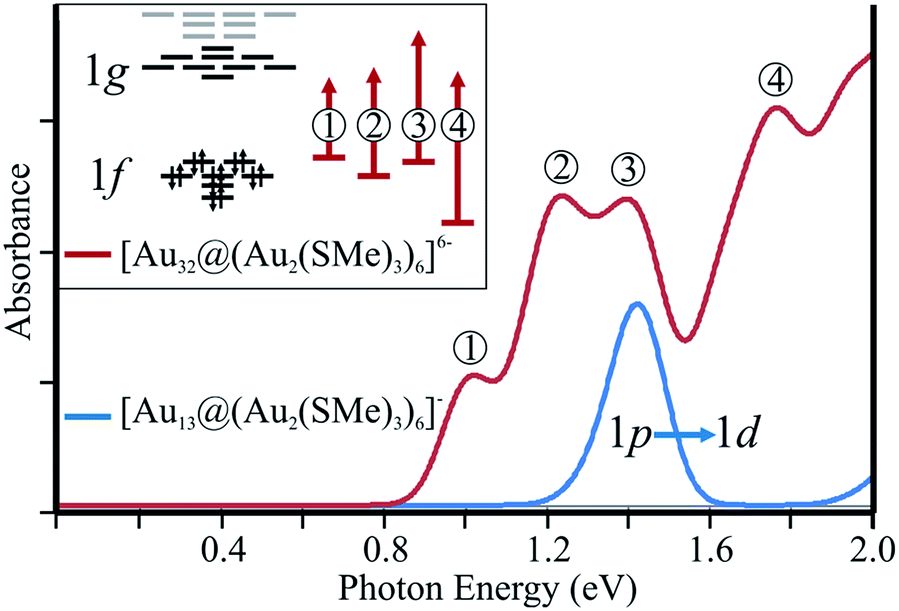 | ||
| Fig. 4 Calculated low-energy absorption spectrum for [Au32@(Au2(SMe)2)6]6−, in comparison to [Au25(SMe)18]−, denoting the nature of each band. | ||
For [Au13@Au12(SR)18]−, the energy of the first electronic transition is calculated to 1.41 eV, which is in close agreement with the recently calculated value of the lowest excitation energy.24 The calculations show that the transition is dominated by a 1p → 1d character. For [Au32@Au12(SMe)18]6−, we obtained several electronic excitation energies in this energy range. The calculations on [Au32@Au12(SMe)18]6− suggest a richer low-energy spectrum with several peaks of which four peaks can readily be characterized. To understand the reasons for the differences between the electronic excitation spectra, further analysis of the transitions can be made in terms of the frontier orbital structure (inset Fig. 4). The first two peaks at 1.01 and 1.23 eV, involve the occupied 1f block and the unoccupied 1g levels of the 1f → 1g transitions. The excitation energy at 1.40 eV is dominated by a 1f → ligand character. The transition character of the last peak in the studied energy region is dominated by a transition between the gold orbitals of the 5d-block and the 1g level. The calculated excitation energy for this transition is 1.76 eV as compared to 2.40 eV for the corresponding the transition of the [Au13@Au12(SR)18]− cluster. Thus, the thiolate-protected 32-ve core has a more complex low-energy spectrum resulting from 1f → 1g allowed transitions. The spin–orbit coupling effects on the electronic absorption spectra have not been studied here.
For comparison, the isoelectronic compact version of [Au13@Au12(SR)18]6− cluster has been studied. Such isomer involves an endohedral Au4 moiety (ESI†), which is located above of 61.3 kcal mol−1 in relation to the hollow isomer. The absorption spectrum is given on Fig. 5. In addition the calculated excitations for [Au36(SMe)24] (ref. 14) are given, in order to account for a more compact central core as given by its FCC-type core similar to a bulk Au (111) lattice. The first few peaks of both 32-ve core clusters are observed at lower energies in relation to [Au36(SMe)24]. Such isomers exhibit a related frontier electronic structure, resulting in a similar pattern for low energy transitions.
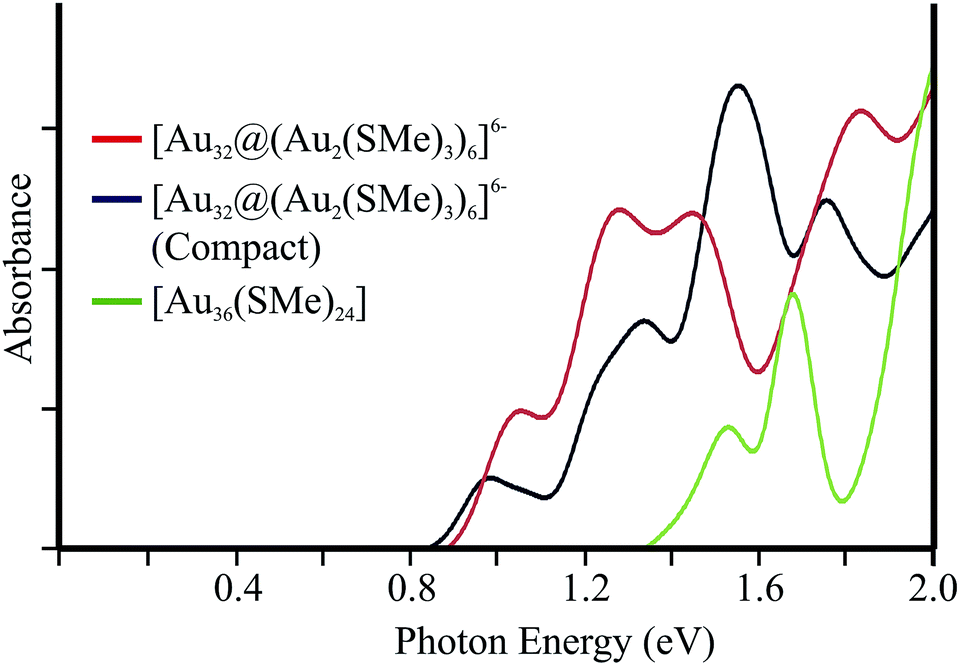 | ||
| Fig. 5 Calculated low-energy absorption spectrum for hollow and compact isomers for [Au32@(Au2(SMe)2)6]6−. In addition, the absorption spectrum of [Au36(SMe)24] is given (see text). | ||
To investigate the nature of the electronic and magnetic properties of the interior of the hollow cluster, the magnetic response (Bind) of an external magnetic field (Bext), given by the shielding tensor (σ) at the center of the cage36,37 was calculated. The calculation of the magnetic shielding inside the Au32 cage provides information about the character and strength of the magnetically induced current density in the shell of molecular cage.20 The calculated magnetic shielding in the center of the cage indicates whether the gold cluster mainly sustains diatropic or paratropic currents, corresponding to a three-dimensional aromatic or antiaromatic character of the gold cage, respectively.33 For the bare Au32 cluster the magnetic shielding calculated at the GIAO PBE-ZORA/TZ2P level is −83.1 ppm, which agree well with the value of −100 ppm as previously obtained at the DFT level using the IGLO method.20 For [Au32@Au12(SMe)18]6−, the magnetic shielding in the center of the molecule is −63.3 ppm, which suggests that the cluster is indeed spherical aromatic. According to the magnetic shielding calculations, the thiolate-protected Au32 cage is somewhat less aromatic than the unsubstituted one. The shieldings are almost independent of the employed DFT functional, since at the BP86-ZORA/TZ2P level the shieldings at the cage center of Au32 and [Au32@Au12(SMe)18]6− are −83.2 ppm and −63.4 ppm, respectively.
Conclusions
In summary, we have computationally studied and compared molecular properties of unsubstituted Au32 and thiolate-protected [Au32@Au12(SMe)18]6− clusters with a hollow 32-ve gold core, which can be stabilized in polar solvents. The calculated low-energy part of the electronic absorption spectrum has several peaks that can serve as fingerprint for distinguishing between hollow and filled structures of small gold nanoclusters. The absorption character of the hollow structures are dominated by 1f → 1g transitions, which significantly differs from the excitation character of other nanoclusters like [Au25(SMe)18]−, whose low-lying excited states are characterized by 1p → 1d transitions. The calculations show that the addition of the external thiolate protected layer does not significantly alter the molecular structure, electronic properties, and aromatic properties of the Au32 cage, suggesting that even larger golden fullerene structures derived from the Au42, Au72, and Au92 cages can be stable.Acknowledgements
This work has been supported by the Academy of Finland through projects (266227 and 275845) and by the FONDECYT grants 1140359 and Project Millennium RC120001. DS thanks the Swedish Cultural Foundation in Finland, Alexander von Humboldt-Foundation, and the Fulbright Foundation for financial support. The computational resources have been provided by CSC — the Finnish IT Center for Science.Notes and references
- Metal Clusters in Chemistry, ed. P. Braunstein, L. A. Oro and P. R. Raithby, Wiley-VCH, Weinheim, Germany, 1999 Search PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- M. Homberger and U. Simon, Philos. Trans. R. Soc., A, 2010, 368, 1405 CrossRef CAS PubMed.
- R. W. Murray, Chem. Rev., 2008, 108, 2688 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157 CrossRef CAS PubMed.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847 RSC.
- P. Jena, J. Phys. Chem. Lett., 2013, 4, 1432 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2010, 131, 2490 CrossRef PubMed.
- R. Jin, Nanoscale, 2015, 7, 1549 RSC.
- T. Ohta, M. Shibuta, H. Tsunoyama, Y. Negishi, T. Eguchi and A. Nakajima, J. Phys. Chem. C, 2013, 117, 3674 Search PubMed; H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280 CrossRef CAS PubMed.
- M. MacDonald, P. Zhang, N. Chen, H. Qian and R. Jin, J. Phys. Chem. C, 2009, 115, 65 Search PubMed; H. Qian and R. Jin, Nano Lett., 2009, 9, 4083 CrossRef CAS PubMed.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206 CrossRef CAS PubMed; C. Zeng, Y. Chen, G. Lia and R. Jin, Chem. Commun., 2014, 50, 55–57 RSC.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035 CAS.
- A. Dass, S. Theivendran, P. R. Nimmala, C. Kumara, V. R. Jupally, A. Fortunelli, L. Sementa, G. Barcaro, X. Zuo and B. C. Noll, J. Am. Chem. Soc., 2015, 137, 4610 CrossRef CAS PubMed.
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef CAS; P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578 CrossRef.
- J. Li, X. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 7, 864 CrossRef PubMed.
- M. P. Johansson, D. Sundholm and J. Vaara, Angew. Chem., Int. Ed., 2004, 43, 2678 CrossRef CAS; M. P. Johansson, J. Vaara and D. Sundholm, J. Phys. Chem. C, 2008, 112, 19311 Search PubMed.
- H. S. De, S. Krishnamurty and S. Pal, Catal. Today, 2012, 198, 106 CrossRef CAS.
- Amsterdam Density Functional (ADF) Code, release 2014, Vrije Universiteit, Amsterdam, The Netherlands, 2014, http://www.scm.com Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. E. Jiang, Nanoscale, 2013, 5, 7149 RSC.
- J. Akola, K. A. Kacprzak, O. Lopez-Acevedo, M. Walter, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2010, 114, 15986 CAS.
- D.-E. Jiang, M. Kühn, Q. Tang and F. Weigend, J. Phys. Chem. Lett., 2014, 5, 3286 CrossRef CAS PubMed.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS; K. G. Dyall and K. Fægri, Introduction to Relativistic Quantum Chemistry, Oxford University Press, New York, 2007 and references therein Search PubMed.
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972 CrossRef CAS.
- R. van Leeuwen and E. J. Baerends, Phys. Rev. A, 1994, 49, 2421 CrossRef CAS.
- Y. Niihori, C. Uchida, W. Kurashigea and Y. Negishi, Phys. Chem. Chem. Phys., 2016, 18, 4251 RSC.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399 CrossRef CAS PubMed; H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 Search PubMed.
- I. Chakraborty, A. Govindarajan, J. Erusappan, A. Ghosh, T. Pradeep, B. Yoon, R. L. Whetten and U. Landman, Nano Lett., 2012, 12, 5861 CrossRef CAS PubMed.
- M. Rauhalahti and A. Muñoz-Castro, RSC Adv., 2015, 5, 18782 RSC.
- D. Manna, T. Jayasekharan and T. K. Ghanty, J. Phys. Chem. C, 2013, 117, 18777 CAS; T. Jayasekharan and T. K. Ghanty, J. Phys. Chem. C, 2010, 114, 8787 Search PubMed.
- T. Heine, C. Corminboeuf and G. Sifert, Chem. Rev., 2005, 105, 3889 CrossRef CAS PubMed.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2001, 40, 2834 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dodecahedral and icosahedral motifs in Au32, comparison between the absorption spectrum obtained at the PBE and LB94 functionals for [Au25(SMe)18]−, and compact isomer of the studied cluster. See DOI: 10.1039/c5ra27683g |
| This journal is © The Royal Society of Chemistry 2016 |