
Surface oxygen vacancy assisted electron transfer and shuttling for enhanced photocatalytic activity of a Z-scheme CeO2–AgI nanocomposite†
M. Jahurul Islam,
D. Amaranatha Reddy,
Jiha Choi and
Tae Kyu Kim*
Department of Chemistry and Chemical Institute for Functional Materials, Pusan National University, Busan 609-735, Republic of Korea. E-mail: tkkim@pusan.ac.kr
First published on 12th February 2016
Abstract
Surface-oxygen-vacancy-promoted Z-scheme CeO2–AgI heterostructured photocatalysts were successfully fabricated via a hydrothermal route combined with a precipitation process. Surface oxygen vacancies were formed on the synthesized CeO2–AgI photocatalyst, as determined by X-ray photoelectron spectroscopy. These oxygen vacancies could extend the lifetime of the charge carriers and enhance the photocatalytic activity of these catalysts for rhodamine B (RhB) dye degradation. Among the as-synthesized photocatalysts, the 20 wt% CeO2–AgI (CA-2) nanocomposite demonstrated the highest photocatalytic activity towards the degradation of RhB with 3.28- and 29.8-fold higher activity than pure AgI and CeO2 nanostructures, respectively. In addition, to ensure the visible light photocatalytic activity of the CeO2–AgI nanocomposite, decomposition studies were performed using a colorless substrate such as phenol. The mechanism for the enhanced photocatalytic performance of the CeO2–AgI photocatalyst is proposed to be based on efficient separation of photogenerated electron–hole pairs through a Z-scheme system, in which oxygen vacancy states promote charge separation. Experiments using scavengers of reactive species combined with photoluminescence analysis provide significant evidence for the oxygen-vacancy-mediated Z-scheme mechanism of the photocatalyst. Moreover, the as-prepared oxygen-deficient CeO2–AgI photocatalysts exhibited excellent cycling stability.
Introduction
Photocatalysis using safe, sustainable, and inexhaustible solar energy has been widely used in water purification to remove organic pollutants, which include organic dyes, pesticides, and herbicides.1 Over the past few years, considerable efforts have been made towards the development of highly efficient and robust photocatalysts for the oxidative degradation of organic dyes to harmless end-products such as CO2, H2O, and NH3.2 Recently, semiconductor nanostructures have gained traction as efficient and cost-effective photocatalysts for water purification applications. These nanostructures have typically been constructed using wide band gap semiconducting oxides such as TiO2 (band gap ∼ 3.2 eV), which possess exceptional chemical stability and a strong resistance to corrosion under the operating conditions. However, due to their large band gaps, these TiO2-based catalysts require UV activation, which limits their efficiency under solar irradiation.3 Therefore, the design and synthesis of solar light-active photocatalysts with an optimum balance between stability and efficiency remains an enduring challenge in this rapidly developing field.Recently, extensive research efforts have focused on the development of plasmonic noble metal photocatalysts for water remediation. Of these, silver halides (AgX, X = Cl, Br, I) are considered to be viable alternatives to TiO2 photocatalysts and have been extensively employed for photocatalytic oxidations.4 Among these Ag-based photocatalysts, AgI has shown promising efficiency for decomposition of organic dyes by sunlight irradiation.5 However, the stability of these catalysts is insufficient for extended reuse, since AgI photocatalysts are completely destroyed upon recycling due to the formation of metallic Ag.6 Current approaches to this problem have centered on the development of heterostructured nanocomposites as photocatalysts. These heterostructures have suitable band positions for the facile transfer of photogenerated electrons formed in the conduction band (CB) of the AgI nanostructures to the supporting semiconductor nanostructure partners, so that the photogenerated carriers are efficiently separated. This efficient electron migration from AgI to the supporting semiconductor nanostructures promotes the stability of AgI nanostructures and enhances their photocatalytic performance.7 Following this strategy, researchers have recently made significant advances in generating various nanohybrid systems containing a variety of semiconductors such as Bi2WO6,8 YVO4,9 TiO2,10 and Ag3PO4.11 These nanohybrid systems exhibit exceptional catalytic stability and superior performance in the photo-oxidation of organic dyes. Moreover, in comparison to conventional supporting semiconductors, oxygen-vacancy-rich materials are more efficient in preserving their intrinsic crystal structures, which contributes to enhanced photocatalytic performance under visible light irradiation.12 Therefore, heterostructure formation with oxygen-vacancy-rich semiconductor supports is an important strategy to improve the efficiency of charge carrier separation and enhance the solar light-driven photocatalytic activity of AgI nanostructures. Herein, we introduce oxygen-vacancy-rich CeO2 nanostructures to immobilize AgI nanostructures towards the synthesis of CeO2–AgI heterostructure nanocomposites.
Cerium(IV) oxide, commonly known as ceria, has generated wide interest due to its unique chemical, mechanical, and magnetic properties originating from its two oxidation states. It has found wide application in catalysis where it plays the roles of both a catalyst support and a catalyst.13,14 Surface oxygen vacancies on ceria are expected to promote efficient catalytic metal particle dispersion when CeO2 is used as a support. Under reducing conditions, the reduction of neighboring Ce4+ ions to Ce3+ occurs, which facilitates oxygen vacancy formation on the surface. These Ce3+ and oxygen vacancies can act as active sites for many catalytic reactions.15 Hence, the generation of surface-oxygen-vacancy-rich CeO2 and its use in the synthesis of CeO2–AgI nanostructures is a preferred strategy for improving the catalytic activity of AgI photocatalysts.
Z-Scheme photocatalysts have generated considerable interest among researchers due to their strong redox capability and high photocatalytic activity compared to single-component band-to-band transfer photocatalysts.16 These Z-scheme systems are usually obtained by coupling two different photocatalysts to yield a direct Z-scheme system.17 In some cases these coupled photocatalysts are combined with an appropriate intermediate (e.g. noble metals).18 The more negative CB and more positive valance band (VB) potentials in Z-scheme photocatalysts contribute significantly to improved photocatalytic performance by effective separation of photogenerated electron–hole pairs.19 The mediator-free direct Z-scheme photocatalytic system exhibits high stability and is economically more feasible for practical applications in comparison to the mediator-containing ternary Z-scheme system.20,21 In this respect, oxygen-vacancy-rich CeO2 nanostructures have tremendous potential to be used as self-mediators in highly efficient and stable Z-scheme photocatalytic systems with two different semiconductors. To the best of our knowledge, the construction of Z-scheme CeO2–AgI heterostructures with vastly improved photocatalytic activity and photostability has not previously been studied.
Herein, we report a systematic study of CeO2–AgI as a photocatalyst for organic dye degradation based on Z-scheme phenomena. A series of physical and chemical analyses uncovered the factors that characterize CeO2–AgI nanocomposites as an efficient surface oxygen vacancy-mediated Z-scheme photocatalyst. We believe that this study will play a significant role in guiding the design and synthesis of novel surface oxygen vacancy-induced Z-scheme photocatalysts with high photocatalytic activity and photostability.
Results and discussion
Morphological characterization of photocatalysts
The morphologies of the as-prepared AgI and CeO2–AgI nanocomposites were characterized by field-emission scanning electron microscopy (FESEM) and are presented in Fig. 1. As shown in Fig. 1(a), the pure AgI consists of inhomogeneous nanoparticles with various sizes (<100 nm) and their aggregates (2–3 μm), which may have formed by agglomeration of the primary AgI nanoparticles. When nanosized CeO2 was introduced to the pure AgI for the preparation of CeO2–AgI nanocomposites, the spherical AgI nanostructures were wrapped with a nanoplate morphology of CeO2 nanoparticles. The FESEM image (Fig. 1(b)) clearly shows that CeO2–AgI nanocomposites contain two distinct phases, which indicates that CeO2 nanostructures are present on the surface of AgI. For a more detailed analysis, both the pure AgI and CeO2–AgI nanocomposite were analyzed by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM), and the results are shown in Fig. 1(c–f). The TEM images clearly show a uniform distribution of CeO2 nanocrystals on the AgI nanosphere surface (Fig. 1(e) and S1(f)†). The HRTEM images of the CeO2–AgI nanocomposite more clearly show two sets of lattice fringes. The lattice fringe with a d-spacing of 0.31 nm is assigned to the (111) plane of cubic CeO2, whereas the lattice fringe of 0.37 nm corresponds to the (002) plane of AgI. X-ray energy dispersive spectroscopy (EDS) elemental mapping of the samples was carried out to further analyze the distribution and spatial homogeneity of Ag, I, Ce, and O elements (Fig. S1(a–e)†). These elements are uniformly dispersed in specific regions of the CeO2–AgI nanostructures. The Ag and I elements are present in the bulk of the material with a uniform lateral intensity, while the Ce and O elements are distributed on the outer surface of the nanospheres. In addition, a detailed chemical analysis of CeO2, AgI, and CeO2–AgI nanocomposites was conducted by EDS (Fig. S2(a–c)†). The EDS spectrum of AgI shows peaks belonging to Ag and I without any impurities. The CeO2–AgI nanocomposite contains Ce, O, Ag, and I, whereas pure CeO2 shows only Ce and O peaks. This result further confirms that CeO2 nanoparticles are anchored on the AgI nanostructure. | ||
| Fig. 1 FESEM images of AgI (a) and CeO2–AgI composite (b), TEM (c) and HRTEM (d) images of AgI, TEM (e) and HRTEM (f) images of CeO2–AgI composite. | ||
XRD analysis of the photocatalysts
Crystallographic structures and orientations of the as-fabricated CeO2–AgI nanocomposite, AgI, and pure CeO2 were investigated by X-ray diffraction (XRD) and the results are displayed in Fig. 2(a). The XRD patterns of bare AgI have been indexed to the hexagonal phase of β-AgI (JCPDS No. 09-0374) with six obvious characteristic diffraction peaks at 2θ ≈ 22.48°, 23.88°, 25.44°, 39.36°, 42.71°, and 46.44° corresponding to the (100), (002), (101), (110), (103), and (112) planes, respectively. In addition to these peaks, very small diffraction peaks at around 56.85° and 62.42° are evident, which are ascribed to the (400) and (331) planes, respectively, of γ-AgI. In the as-synthesized nanocomposites, β-AgI and γ-AgI phases coexist, as observed from the results.22,23 The sharp and intense diffraction peaks suggest that the as-synthesized AgI and CeO2–AgI nanostructures have a crystalline nature. Moreover, no crystalline impurities related to reaction by-products, such as Ag metal or Ag clusters, are observed, which indicates high purity of the product. The average nanocrystallite sizes (D) of the AgI and CeO2–AgI nanostructures were estimated using the Debye–Scherrer formula (D = 0.89λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where λ is the wavelength of the Cu Kα irradiation, β is the full width at half maximum (FWHM) of the diffraction peak, and θ is the diffraction angle for the (002) planes of hexagonal β-AgI). The calculated D values are in the range of 40–80 nm. In the case of the CeO2 nanoparticles, all the diffraction peaks can be indexed as the cubic fluorite-type CeO2 structure (JCPDS No. 81-0792)24 with the strongest peak (111) at 2θ = 28.67°. The calculated average crystallite size (D) of pure CeO2 nanoparticles is around 51 nm. The XRD patterns of the CeO2–AgI nanocomposites clearly exhibit two sets of diffraction peaks, which fully correspond to polycrystalline structures of CeO2 and AgI phases, indicating that the CeO2–AgI nanostructure was successfully prepared. Furthermore, the intensities of the CeO2 diffraction peaks in the CeO2–AgI nanocomposites gradually increase with an increase in the wt% of CeO2. This result is consistent with the FESEM and TEM studies.
cosθ, where λ is the wavelength of the Cu Kα irradiation, β is the full width at half maximum (FWHM) of the diffraction peak, and θ is the diffraction angle for the (002) planes of hexagonal β-AgI). The calculated D values are in the range of 40–80 nm. In the case of the CeO2 nanoparticles, all the diffraction peaks can be indexed as the cubic fluorite-type CeO2 structure (JCPDS No. 81-0792)24 with the strongest peak (111) at 2θ = 28.67°. The calculated average crystallite size (D) of pure CeO2 nanoparticles is around 51 nm. The XRD patterns of the CeO2–AgI nanocomposites clearly exhibit two sets of diffraction peaks, which fully correspond to polycrystalline structures of CeO2 and AgI phases, indicating that the CeO2–AgI nanostructure was successfully prepared. Furthermore, the intensities of the CeO2 diffraction peaks in the CeO2–AgI nanocomposites gradually increase with an increase in the wt% of CeO2. This result is consistent with the FESEM and TEM studies.
 | ||
| Fig. 2 (a) XRD patterns of AgI, CeO2, and CeO2–AgI composites with different CeO2 contents; XPS spectra of 20 wt% CeO2–AgI composite: (b) survey spectra, (c) Ag 3d, (d) I 3d, (e) Ce 3d, and (f) O 1s narrow scan spectra. | ||
XPS analysis of the nanocomposites
XPS analysis was performed on the CeO2, AgI, and CeO2–AgI nanocomposite samples to determine the elemental compositions and oxidation states of the respective elements present in the composite. The XPS survey scan confirms the presence of Ag, I, Ce, and O elements in the CeO2–AgI nanocomposites, as shown in Fig. 2(b). XPS survey scans for pure CeO2 and AgI nanocomposites were also performed, as shown in Fig. S3(a) and (d).† The high-resolution XPS spectra of Ag 3d, I 3d, Ce 3d, and O 1s for the CeO2–AgI nanocomposites are shown in Fig. 2(c)–(f). As demonstrated in Fig. 2(c), the Ag 3d spectrum has two peaks at 373.9 and 367.8 eV, which can be assigned to the Ag 3d3/2 orbitals and Ag 3d5/2, respectively, corresponding to the binding energy of Ag+. Similarly, two peaks, Ag 3d3/2 (372.9 eV) and Ag 3d5/2 (366.9 eV), are observed in the XPS spectra of AgI (Fig. S3(a) and (b)†). These findings indicate that no metallic Ag0 appears in either the CeO2–AgI or the AgI. The high-resolution (I 3d) XPS spectrum of CeO2–AgI nanocomposites (Fig. 2(d)) shows two peaks at 630.8 and 619.3 eV, which can be ascribed to I 3d3/2 and I 3d5/2, respectively, in the form of I− ions. The XPS I 3d scan also shows similar peaks, as presented in Fig. S3(c).† These values are consistent with the results published in a previous report.5,25 The Ce 3d peaks can be confirmed by XPS analysis, as shown in Fig. 2(e) and S3(e)† for the CeO2–AgI nanocomposite and CeO2, respectively. CeO2–AgI and CeO2 reveal the mixed valance states of both Ce3+ and Ce4+ owing to its non-stoichiometric nature and multiple d-splitting such as 3d3/2 and 3d5/2. In the CeO2–AgI nanocomposite, the main characteristic peaks of Ce4+ 3d3/2 and Ce4+ 3d5/2 occur at 916.7 and 898.4 eV, respectively, whereas the peaks located at 901.1 and 882.6 eV are assigned to Ce3+ 3d3/2 and Ce3+ 3d5/2, respectively. In addition, three satellite peaks (referred to as ‘shake up’) are located at 907.6 eV for Ce3+ 3d3/2 and at 885.6 and 889.4 eV for Ce3+ 3d5/2, respectively. In the case of CeO2, all the d-splitting peaks are assigned (Ce4+ 3d3/2, Ce4+ 3d5/2, Ce3+ 3d3/2, and Ce3+ 3d5/2 peaks are located at 916.5, 898.3, 901.1, and 882.3 eV, respectively). Three satellite peaks are also assigned at 907.1 eV for Ce3+ 3d3/2 and at 885.1 and 888.5 eV for Ce3+ 3d5/2, respectively. These spectral data are fully consistent with previous reports.26,27The O 1s XPS spectrum of the CeO2–AgI nanocomposites (Fig. 2(f)) shows asymmetric profiles. Generally, the low binding energy of the O 1s peak is ascribed to lattice oxygen, whereas the high binding energy peak is assigned not only to oxygen vacancies but also to surface adsorbed oxygen, hydroxyl, and carbonate groups. Specifically, the O 1s peak can be deconvoluted into three peaks, which show different types of oxygen species, such as lattice oxygen (OL), oxygen vacancies or defects (OV), and chemisorbed or dissociated oxygen (OC) at the binding energies of 529.3, 531.4, and 532.9 eV, respectively.28,29 In the case of CeO2, however, the high resolution O 1s XPS spectrum shows two peaks at binding energies of 529.3 and 531.9 eV, which correspond to OL and OC (Fig. S3(f)†). The observed significant difference between the XPS O 1s spectra of CeO2–AgI and CeO2 could be one cause for the higher photocatalytic activity of the CeO2–AgI nanocomposite than CeO2.29 The photocatalytic activity of the CeO2–AgI nanocomposite depends on the varying oxygen defect concentration and the type of defects. Moreover, the oxygen species from the CeO2–AgI nanocomposite at around 532.9 eV is caused by surface hydroxyl groups on the CeO2–AgI nanocomposite. These hydroxyl groups enhance the trapping of photoinduced electrons and holes; thereby increasing the efficiency of the photocatalytic degradation process.
FTIR measurement
To investigate the chemical interactions of polyvinyl pyrrolidone (PVP) or its fragments with AgI, CeO2, and CeO2–AgI nanostructures, Fourier transform infrared (FTIR) spectra were recorded using the potassium bromide (KBr) pellet method. Typical FTIR spectra of PVP, Ag, CeO2, and CeO2–AgI nanostructures are displayed in Fig. 3(a). The broad absorption peak around 3441 cm−1 in Fig. 3(a) is characteristic of O–H stretching vibrations of hydrogen-bonded water present in the surface hydration layers of the sample. This peak is difficult to avoid in the KBr pellet method due to the extremely hygroscopic nature of the KBr matrix.30 The absorption bands at 2955 cm−1 corresponding to the sp3 C–H stretching vibration31 whereas the characteristic intense peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations can be observed at 1660 cm−1 in the FTIR spectrum of PVP.32 The spectral band situated at 1435 cm−1 corresponds to the CH2 bending vibration while the peak at 1270 cm−1 is attributed to the C–N stretching vibration of the PVP aromatic ring. The CO, C–H, and C–N vibrational peaks that arise in AgI and the CeO2–AgI nanocomposite are shifted slightly towards lower frequencies compared to pure PVP vibrations. This effect might be due to weak interactions between PVP polymer molecules and metal ions (Ag+, Ce3+, Ce4+).33 The peak located at 2350 cm−1 is attributed to the adsorption of CO2 molecules.33,34 In the pure CeO2, additional bands in the region of 1200–1720, 3660–3800 cm−1, and below 800 cm−1 appear due to the stretching frequency of Ce–O. In the CeO2–AgI nanocomposite, all these band peaks appear due to stretching vibrations of Ce–O and Ag–I, confirming the successful formation of the nanocomposite.26,35 Previous work also reported the FTIR absorption band of Ag–I bond around 390–560 cm−1, which is consistent with our result.36 The above results indicate that the PVP polymer stabilizes the nanostructures through steric effects as well as electrostatic interactions between the metal ions and polar functional groups present on the polymer.33
O stretching vibrations can be observed at 1660 cm−1 in the FTIR spectrum of PVP.32 The spectral band situated at 1435 cm−1 corresponds to the CH2 bending vibration while the peak at 1270 cm−1 is attributed to the C–N stretching vibration of the PVP aromatic ring. The CO, C–H, and C–N vibrational peaks that arise in AgI and the CeO2–AgI nanocomposite are shifted slightly towards lower frequencies compared to pure PVP vibrations. This effect might be due to weak interactions between PVP polymer molecules and metal ions (Ag+, Ce3+, Ce4+).33 The peak located at 2350 cm−1 is attributed to the adsorption of CO2 molecules.33,34 In the pure CeO2, additional bands in the region of 1200–1720, 3660–3800 cm−1, and below 800 cm−1 appear due to the stretching frequency of Ce–O. In the CeO2–AgI nanocomposite, all these band peaks appear due to stretching vibrations of Ce–O and Ag–I, confirming the successful formation of the nanocomposite.26,35 Previous work also reported the FTIR absorption band of Ag–I bond around 390–560 cm−1, which is consistent with our result.36 The above results indicate that the PVP polymer stabilizes the nanostructures through steric effects as well as electrostatic interactions between the metal ions and polar functional groups present on the polymer.33
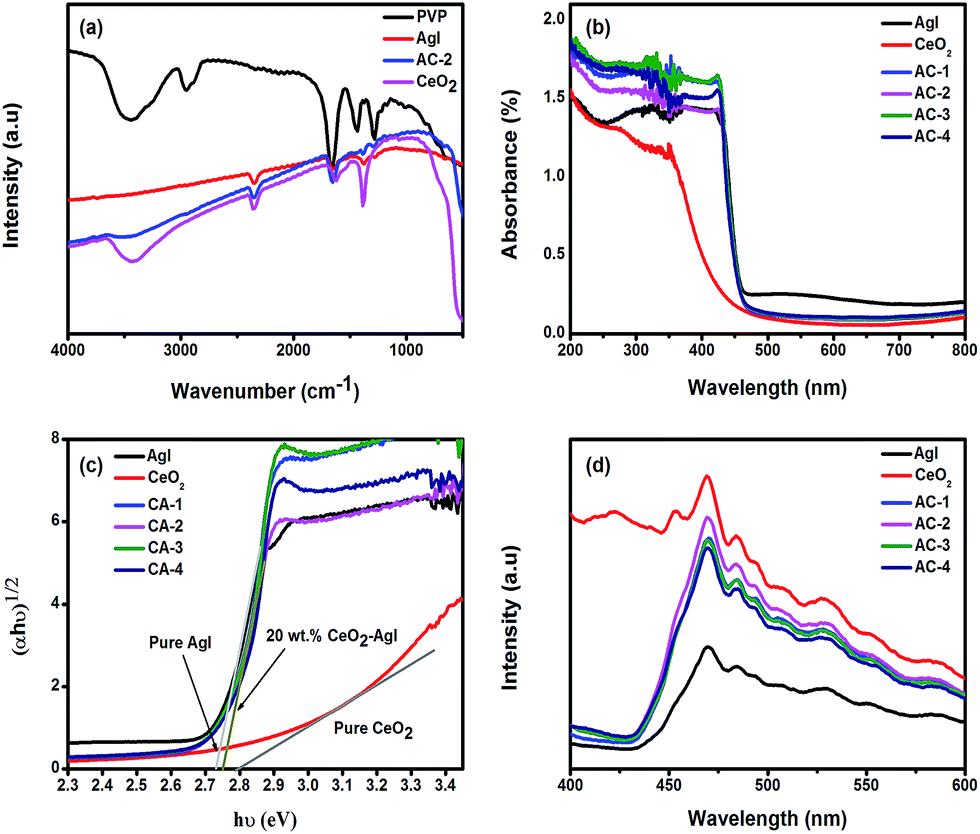 | ||
| Fig. 3 (a) FTIR spectra of PVP, AgI, CeO2, and CeO2–AgI nanocomposite; (b) UV-Vis diffuse-reflectance spectra; (c) optical conductivity of different photocatalysts; and (d) PL spectra of AgI, CeO2, and CeO2–AgI nanocomposite. | ||
UV-Vis absorption spectra analysis and band gap calculation
Optical band gaps of the CeO2–AgI nanocomposites, AgI, and pure CeO2 are displayed in Fig. 3(b). The optical absorption edges of pure AgI were determined using UV-Vis diffuse reflectance spectroscopy (DRS). The resulting CeO2 spectra were estimated from the DRS absorption spectra to be 465 and 450 nm. However, the absorption band edges of the nanocomposites shift slightly towards shorter wavelengths. All CeO2–AgI nanocomposites absorb UV-visible light at higher intensities compared to bare AgI and CeO2. CeO2 shows a lower absorption in the visible region of the spectrum, a feature attributed to ligand-to-metal charge transfer (LMCT) from O2− (2p) to Ce4+ (4f) orbitals.37The energy levels and band gap of the nanocomposite materials play a vital role in determining their optoelectronic properties. The band gap energy of a nanostructure can be calculated using the following formula:
| αhν = A(hν − Eg)(n/2) |
Photoluminescence (PL) analysis
To understand the fate of the photogenerated electron–hole pairs, room temperature photoluminescence (PL) emission spectra of AgI, CeO2, and CeO2–AgI nanocomposites were analyzed in the range of 400 to 600 nm, as shown in Fig. 3(d), with an excitation wavelength of 325 nm. From the figure, it is clear that the AgI nanostructure shows a strong emission band centered at 470 nm, which originates from a direct electron–hole recombination process.40 The PL peaks above 470 nm arise probably from surface defects present on the AgI nanoparticles.41 In addition, the PL intensity of the CeO2–AgI composite is much higher than that of pure AgI. The intensity also depends on the ratio of CeO2 present in the nanocomposite: the intensity is found to increase as the CeO2 amount increases from 10 to 20 wt%. However, the PL intensity decreases at a higher wt% of CeO2, with the highest PL intensity recorded for the nanocomposite samples with 20 wt% of CeO2. In general, a higher PL intensity indicates a higher recombination rate of photoexcited electrons and holes, which has a detrimental effect on the photocatalytic activity. In contrast, a lower PL intensity indicates a lower recombination rate, suggesting that more photoexcited holes and electrons can participate in the redox reactions, thereby enhancing the photocatalytic performance of the material.42 However, in our case, under the experimental conditions, a higher PL intensity indicates a higher photocatalytic activity (please refer to the section on photocatalytic performance). The reason for this contradictory observation may be the presence of surface defects or oxygen vacancies in the CeO2–AgI nanocomposites. At higher concentrations of Ce3+ states and a corresponding high concentration of oxygen vacancy states, a greater percentage of valence electrons can be excited to the defect state or oxygen vacancy states, which would lead to a higher PL signal intensity.43 The presence of these oxygen vacancies was also confirmed by our XPS results (Fig. 2(f)). It has been reported that oxygen vacancies can serve as electron traps, which will reduce the recombination of photogenerated electrons and holes, thereby promoting photocatalytic activity.28 The PL emission peaks of bare CeO2 occur at 422, 452, 468, 484, 495, 508, 528, 553, and 585 nm. These results are in agreement with a previous report on this material.44 The broad violet to blue band at 422 nm originates from the charge transitions from the 4f band to the VB of CeO2. The blue (452 and 468 nm) and weak blue-green emissions (484 and 495 nm) are possibly due to surface defects of CeO2.45 On the other hands, Li et al. reported that the broad emission band around 350–700 nm is responsible for Ce3+ ions and oxide defect in CeO2.46 The broad green band at 528 nm may be attributed to the oxygen vacancies.47 The emission bands ranging from 400 to 500 nm for CeO2 nanoparticles are attributed to the different defect levels ranging from the Ce 4f to O 2p bands.48 Based on the above results, it may be concluded that oxygen vacancies present in the nanostructures act as active centers for electron trapping, and therefore CeO2–AgI composites exhibit both a high intensity in the PL emission spectra and an increase in photocatalytic activity.49Photocatalytic performance
The photocatalytic decolorization of rhodamine B (RhB) was carried out to evaluate the photocatalytic performance of the as-prepared CeO2–AgI nanocomposite under simulated solar light irradiation. The results of these experiments are shown in Fig. S4(a)–(f).† Before the photocatalytic degradation experiment, the RhB dye and suspended photocatalyst solution were stirred together for 30 min in the dark at room temperature to reach an adsorption–desorption equilibrium, which is crucial for optimum surface coverage. Among the photocatalysts (CeO2, AgI, and CeO2–AgI nanocomposites), the CeO2–AgI nanocomposite shows the highest adsorption efficiency (Fig. S5†).50,51 A control experiment confirms that RhB dye degrades very slowly (within 70 min) in the absence of the photocatalyst. The photodegradation process was evaluated by monitoring the change in RhB concentration. The absorption spectra of RhB with the CeO2–AgI nanocomposite photocatalyst under solar light irradiation clearly show that the characteristic absorption peaks corresponding to RhB decrease rapidly as the irradiation time increases, indicating rapid RhB decomposition. The digital images of the color changes occurring during the experiments are presented in Fig. S6.† The shift in absorption to shorter wavelengths (from 553.8 to 496 nm) observed in the presence of the CeO2–AgI nanocomposite suggests that the RhB degradation process occurs by the stepwise removal of the four N-ethyl groups in the dye (successively yielding N,N,N1-triethylated rhodamine, N,N1-diethylated rhodamine, N-ethylated rhodamine, and rhodamine).52 These changes in the chromophore structure during the degradation process are reflected in the decrease in intensities of characteristic peaks and corresponding color changes of the dye solution from pink to yellow, green, and then finally to colorless.52After the adsorption process under dark conditions, the degradation efficiencies of RhB over pure AgI, CeO2, and the CeO2–AgI nanocomposite photocatalysts under simulated solar-light irradiation are shown in Fig. 4(a), which are the normalized concentrations of RhB dye (Ct/C0) as a function of light irradiation time. C0 and Ct denote the initial concentration after the adsorption–desorption equilibrium under dark conditions and the concentration of RhB at different irradiation times (t), respectively. Consequently, the lower Ct/C0 indicates the higher degradation of RhB dye. As shown in Fig. 4(a), the CeO2–AgI nanocomposite photocatalyst shows higher photocatalytic activity than pure AgI and CeO2. After 70 min of irradiation, among the nanocomposites with different CeO2 ratios, the 20 wt% CeO2–AgI (CA-2) exhibits the highest photocatalytic activity for the decolorization of RhB. The degradation efficiencies of RhB over 10, 20, 30, and 40 wt% CeO2–AgI are 82.32, 98.99, 92.93, and 85.35%, respectively. As shown in Fig. S7,† the kinetics for RhB photocatalytic decolorization with the photocatalysts using the equation of ln(Ct/C0) = kt, indicate that the photocatalytic degradation of RhB over these photocatalysts follows a pseudo-first-order kinetics model.41,53,54 For the RhB dye degradation, CeO2 and AgI show relatively slow degradation rates, with k = 1.84 × 10−3 and 1.67 × 10−2 min−1, respectively. The reaction rate constant (k) in the case of other composites, such as CA-1, CA-2, CA-3, and CA-4, show relatively higher degradation rates, with k = 2.18 × 10−2, 5.48 × 10−2, 3.35 × 10−2, and 2.09 × 10−2 min−1, respectively. The 20-wt% CeO2–AgI nanocomposite, which has the best photocatalytic efficiency, shows ∼3.28- and 29.8-fold higher activity than those of AgI and CeO2, respectively. Similarly, Gu et al. reported RhB degradation by CeO2 nanoparticle sensitized with CdS nanorod photocatalyst with the rate constant of 0.0700 min−1.55 Overall, these results demonstrate that the photocatalytic efficiency of the 20 wt% CeO2–AgI nanocomposite with respect to RhB degradation is much higher than not only those of pure AgI and CeO2, but also the other CeO2–AgI nanocomposites and pure TiO2 under simulated solar light irradiation. Considering the FESEM (Fig. 1(b)) and XRD (Fig. 2(a)) results, the higher photocatalytic activity of the 20 wt% CeO2–AgI nanocomposites can be attributed to a more homogeneous dispersion in the catalyst nanostructure than in the case of the 40 wt% CeO2–AgI photocatalyst, while the lower photocatalytic activity of the 10 wt% CeO2–AgI photocatalyst could be due to a limited amount of CeO2 phase present. In the photocatalytic assessments of Bi2O3/CeO2 nanocomposites, photocatalytic efficiency also depends on the appropriate cavities as well as the particle size of the nanospheres.56 Additionally, the differences in the photocatalytic performance of the pure AgI and CeO2–AgI photocatalysts could also depend on the presence of oxygen vacancies or surface defects in the nanocomposites. However, it is quite clear that a homogeneous dispersion of CeO2 on the AgI nanostructure is an important requirement for the preparation of highly efficient solar light-driven photocatalysts. This inference is consistent with previous studies and reinforced by our results.6,57
 | ||
| Fig. 4 (a) Photocatalytic degradation of RhB by CeO2, AgI, CeO2–AgI composites, and TiO2 under simulated solar light; (b) recycling of 20 wt% CeO2–AgI composite; (c) scavenger effects on the degradation of aqueous RhB solution with 1 mM scavenger at 70 min light irradiation time. | ||
Dye sensitization of photocatalysts is a significant factor that could affect photocatalytic performance. To exclude the contribution of RhB dye sensitization to catalysis under visible light, colorless phenol was chosen as a model compound to evaluate the photocatalytic performance of the CeO2–AgI nanocomposite. Fig. S8(a)† shows the normalized absorption spectra changes of an aqueous solution of phenol (10 mg L−1) with 100 mg of the 20 wt% CeO2–AgI photocatalyst under solar light irradiation as a function of light irradiation time. As in the RhB photocatalytic measurements, before the photocatalytic experiment, the phenol solution containing the photocatalyst was magnetically stirred for 30 min in the dark at room temperature to achieve adsorption–desorption equilibrium. As shown in Fig. S9,† the adsorption efficiency of phenol onto the CeO2–AgI photocatalyst was found to be less than that of RhB dye. The characteristic absorption peak of phenol at 269.9 nm was chosen for monitoring for the progress of the photocatalytic degradation process. From Fig. S8(a and b),† it can be clearly seen that the absorption peaks corresponding to the phenol concentration decrease with the increase in irradiation time, indicating the rapid phenol decomposition. The degradation of phenol with AgI and 20 wt% CeO2–AgI photocatalysts under simulated solar light irradiation is shown in Fig. S8(c).† The photocatalytic study of Ag3PO4/CeO2 nanocomposite showed similar results for the phenol degradation, where Ag3PO4/CeO2 nanocomposite exhibits a higher activity compared to the pure Ag3PO4 nanostructure.24 The phenol concentration clearly decreases with irradiation time. Furthermore, the 20 wt% CeO2–AgI nanocomposite photocatalyst shows better photocatalytic performance for phenol degradation than the pure AgI photocatalyst. This outcome with phenol degradation provides powerful evidence to show that the photocatalytic activity of the 20 wt% CeO2–AgI nanocomposite is inherent to the catalyst, with minimal contribution from dye sensitization under visible light irradiation.
Reusability and stability
Reusability and stability of the nanocomposite photocatalysts are significant parameters that have a bearing on the practical utility of these catalysts. To estimate these parameters for the as-prepared CeO2–AgI nanocomposite photocatalyst, recycling reactions were carried out for the degradation of RhB over the 20 wt% CeO2–AgI nanocomposite under simulated solar light irradiation. The results of these studies are displayed in Fig. 4(b). For the recycling process, previously used CeO2–AgI nanocomposite photocatalyst was removed from the solution by centrifugation, dried at 80 °C for 8 h, and then reused for the degradation of the same concentration of RhB dye. Catalyst activity measurements after five consecutive cycles showed excellent stability and recyclability of the CeO2–AgI nanocomposite photocatalyst. The XRD patterns of the 20 wt% CeO2–AgI nanocomposite were also investigated after five recycling runs for photodegradation of RhB, as shown in Fig. S10.† The diffraction patterns obtained before and after the reactions reveal that the phase and structure of the catalysts remain unchanged. These results indicate that the CeO2–AgI nanocomposite heterostructure photocatalyst is sufficiently stable during photodegradation of the organic dye and can be reused over many cycles without significant loss in performance.Proposed mechanism for the photocatalytic performance of CeO2–AgI
The photocatalytic mechanism of dye degradation was investigated for the CeO2–AgI nanocomposite photocatalyst. In general, organic dyes such as RhB are degraded through photocatalytic oxidation processes, which involve a large number of reactive species including h+, ˙O2−, and ˙OH radicals. To fully elucidate the photocatalytic processes in detail, the effect of scavengers on the decolorization (i.e. photo-induced degradation) of RhB should be examined. We have used ethylenediaminetetraacetic acid (EDTA), benzoquinone (BQ), and t-butyl alcohol (TBA) as the scavengers for h+, ˙O2−, and ˙OH radicals, respectively.17,51 From Fig. 4(c), it is clear that the addition of EDTA slightly affects the decolorization rate of RhB over the 20 wt% CeO2–AgI nanocomposite, suggesting that the h+ reactive species are only slightly involved in the photocatalytic process. On the other hand, the degradation efficiency of the RhB dye decreases considerably after the addition of BQ and TBA, which indicates that ˙O2− and ˙OH radicals are the main active species in the photocatalytic decolorization process of RhB under simulated solar light irradiation.The photocatalytic activity is mainly affected by the recombination rate of the photogenerated electrons and holes. In the case of pure AgI, the photoinduced electrons and holes recombine rapidly because of its narrow band gap. However, in the case of the CeO2–AgI nanocomposite, the photoinduced carriers can transfer easily between CeO2 and AgI due to the matching of their band positions. The band edge positions of the CB and VB of the CeO2 and AgI nanostructure can be determined using the following equations:
| EVB = χ − Ee + (0.5)Eg |
| ECB = EVB − Eg |
Based on the scavenger experiments, PL analyses, and calculated band gap structures of CeO2 and AgI, we propose a possible mechanism for the photocatalytic activity of the CeO2–AgI photocatalyst, as outlined in Scheme 1. According to Scheme 1(a), once the photoexcited charge carriers transfer between CeO2 and AgI in the nanocomposite, the photoexcited electrons in the CB of AgI will migrate to the CB of CeO2 and the photoexcited holes in the VB of CeO2 will migrate to the VB of AgI. This process is very common in a large number of nanocomposite photocatalysts. The accumulated electrons in the CB of CeO2 cannot reduce more O2 to yield ˙O2− because the CB edge potential of CeO2 (−0.32 eV vs. normal hydrogen electrode (NHE)) is more positive than the standard redox potential E°(O2/˙O2−) (−0.33 eV vs. NHE).18,58 Furthermore, Ce4+ in CeO2 (4f0 5d0 6s0) would accept an electron at the CB and form Ce3+ (4f1 5d0 6s0), which would generate an oxygen vacancy state slightly below the CB edge potential position.13,15,59 This oxygen vacancy state (OV) is more positive than the standard redox potential E°(O2/O2−) value. As a result, in this oxygen vacancy state, CeO2 cannot reduce O2 to the ˙O2− radical. On the other hand, the VB position of AgI (2.34 eV vs. NHE) is more negative than that of CeO2 (2.47 eV vs. NHE), indicating that the capability of h+ in the VB of AgI to oxidize OH− or H2O to ˙OH radicals is lower than that of CeO2. However, under the experimental conditions, ˙O2− and ˙OH reactive species play a key role in the photocatalytic reaction. The PL intensity of the CeO2–AgI nanocomposite increases and at the same time the nanocomposite shows high photocatalytic activity compared to pure AgI. This unexpected observation cannot be explained by the common electron–hole separation process, as shown in Scheme 1(a). Therefore, this scheme is inadequate for the description of the photocatalytic mechanism of CeO2–AgI nanocomposites.
 | ||
| Scheme 1 A model for the photocatalytic mechanism of the CeO2–AgI nanocomposite with Z-scheme and oxygen vacancy states. | ||
The photoexcited electron–hole separation process is tentatively proposed, as shown in Scheme 1(b). In this process, the photoexcited electron in the CB of CeO2 (4f0 5d0 6s0) immediately reacts with Ce4+ and forms Ce3+ (4f1 5d0 6s0), which is stabilized by creating an oxygen vacancy state.60,61 The electron is then rapidly transferred to the VB of AgI, resulting in the recombination of the electrons in the CB of CeO2 with the photoexcited holes in the VB of AgI. This process leads to the presence of more electrons in the CB of AgI and holes in the VB of CeO2, which participate in the reduction and oxidation reactions, respectively. The electrons in the CB of AgI (−0.39 eV vs. NHE) have more negative potential to rapidly reduce molecular oxygen (O2) to ˙O2−, which then plays a role in the degradation of the RhB dye.62,63 Simultaneously, the holes in the VB of CeO2 (+2.47 eV vs. NHE) have more positive potential to generate plenty of active ˙OH radicals with powerful oxidation potential. As a result, the photocatalytic performance is significantly enhanced and RhB is photocatalytically degraded through the ˙O2−, ˙OH, or direct h+ oxidation pathways. In this proposed mechanism, CeO2 functions as an electronic modulator for the electron-transfer process where the valence and defect structures of CeO2 play an important role. Similar types of oxygen vacancy and Z-scheme mechanism have been reported. Wang et al.12 reported oxygen vacancies involved band gap narrowing and improved the visible light photocatalytic activity of ZnO whereas Bai et al.64 showed both surface oxygen defect and hybridization of graphene enhanced the activity of ZnO as photocatalyst. In addition, it has been shown that the reduction of Ce4+ generates the shallow defect near the CB and subsequently forms the surface oxygen vacancy in CeO2.13,15,59,65 Moreover, it was reported that efficiently separation of photogenerated electron–hole pairs and Z-scheme mechanism enhance the photocatalytic activity of the nanocomposites.16,66,67 These findings also bold our concept to propose the mechanism for the photocatalytic activity of CeO2–AgI nanocomposite. Based on our results and according to the proposed mechanism shown in Scheme 1(b), the favorable separation process of the photogenerated electron–hole pairs of the CeO2–AgI system is characteristic of an oxygen vacancy-driven Z-scheme nanocomposite photocatalyst.
Evidence for the mechanism from PL spectra
As shown in Fig. 3(d), the PL spectra have a strong emission peak at around 468 nm due to oxygen vacancies.45,67 In addition, Ce3+ ions are mainly responsible for blue luminescence centered at 468 nm in CeO2–AgI nanocomposite.68 From the Fig. 3(d) it can be clearly seen that the PL intensities of the CeO2–AgI nanocomposite are higher than that of pure AgI. Additionally, the PL increases with the increase in the amount of CeO2 from 10 to 20 wt% and then decreases at higher CeO2 content. This observation indicates that a higher PL intensity is related to higher photocatalytic activity under our experimental conditions, a result that is contradictory to the accepted theory regarding general PL experiments. The main reason for this apparent contradiction may lie in the recombination process between the photoexcited electrons and holes. If the photoexcited electrons and holes are transferred, as shown in Scheme 1(a), the PL intensities of the CeO2–AgI nanocomposite must be lower than that of pure AgI because the photoexcited electrons and holes of CeO2 and AgI are separated effectively and hence recombine slowly. However, the results show that the PL intensity of the CeO2–AgI composite is much higher than that of pure AgI, which suggests that the photoexcited electrons and holes are not transferred, as shown in Scheme 1(a). In Scheme 1(b), the higher PL intensities of the samples are attributed to the higher recombination rate between photoexcited electrons in the CB of CeO2 and holes in the VB of AgI through oxygen vacancy states. Therefore, the electrons in the CB of AgI with more negative potential, and the holes in the VB of CeO2 with more positive potential, participate in reduction and oxidation reactions to produce ˙O2− and ˙OH radicals, respectively. These processes would improve the photocatalytic activity significantly. Hence, it can be inferred that the CeO2–AgI nanocomposite system is a typical oxygen-vacancy-mediated Z-scheme photocatalyst exhibiting excellent photocatalytic redox performance.Conclusions
Under simulated solar light conditions, high-efficiency oxygen vacancy mediated Z-scheme CeO2–AgI nanocomposite photocatalysts were fabricated via a hydrothermal route combined with a simple precipitation process. The synthesized nanocomposite photocatalyst exhibits excellent photocatalytic performance on not only RhB dye degradation but also on the degradation of colorless phenol. The photocatalytic activity of the CeO2–AgI nanocomposite (20 wt%) was superior to that of pure AgI and CeO2. The excellent photocatalytic activity of the nanocomposite photocatalyst can be attributed to the efficient separation of photogenerated electron–hole pairs through the Z-scheme system of the CeO2–AgI nanocomposite. The surface oxygen vacancy states of CeO2 promote the separation efficiency. The Z-scheme photocatalytic process of the composite photocatalysts was also supported by PL analysis and scavenger experiments. Therefore, based on the aforementioned results of this study, the synthesized CeO2–AgI nanocomposites are expected to be useful as efficient solar light-driven photocatalysts with wide practical applications.Acknowledgements
This work was financially supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MEST and MSIP) (2007-0056095, 2013S1A2A2035406, 2013R1A1A2009575, and 2014R1A4A1001690). This research has been supported in part by Global Research Laboratory Program [Grant No. 2009-00439] and by Max Planck POSTECH/KOREA Research Initiative Program [Grant No. 2011-0031558] through the National Research Foundation of Korea (NRF) funded by Ministry of Science, ICT & Future Planning.Notes and references
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. G. Sun and J. H. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- P. Gao, H. Ma, T. Yan, D. Wu, X. Ren, J. Yang, B. Du and Q. Wei, Dalton Trans., 2015, 44, 773–781 RSC.
- G. Li, Y. Wang and L. Mao, RSC Adv., 2014, 4, 53649–53661 RSC.
- C. Hu, X. Hu, L. Wang, J. Qu and A. Wang, Environ. Sci. Technol., 2006, 40, 7903–7907 CrossRef CAS PubMed.
- H. Yu, L. Liu, X. Wang, P. Wang, J. Yu and Y. Wang, Dalton Trans., 2012, 41, 10405–10411 RSC.
- Z. Zhang, D. Jiang, C. Xing, L. Chen, M. Chen and M. He, Dalton Trans., 2015, 44, 11582–11591 RSC.
- B. Chen, Y. Deng, H. Tong and J. Ma, Superlattices Microstruct., 2014, 69, 194–203 CrossRef CAS.
- K. Vignesh, A. Suganthi, B.-K. Min, M. Rajarajan and M. Kang, RSC Adv., 2015, 5, 576–585 RSC.
- C. An, W. Jiang, J. Wang, S. Wang, Z. Ma and Y. Li, Dalton Trans., 2013, 42, 8796–8801 RSC.
- Z. Chen, W. Wang, Z. Zhang and X. Fang, J. Phys. Chem. C, 2013, 117, 19346–19352 CAS.
- J. Wang, Z. Wang, B. Huang, Y. Ma, Y. Liu, X. Qin, X. Zhang and Y. Dai, ACS Appl. Mater. Interfaces, 2012, 4, 4024–4030 CAS.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- J. Beckers and G. Rothenberg, Green Chem., 2010, 12, 939–948 RSC.
- N. V. Skorodumova, S. I. Simak, B. I. Lundqvist, I. A. Abrikosov and B. Johansson, Phys. Rev. Lett., 2002, 89, 166601–166604 CrossRef CAS PubMed.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- N. Tian, H. Huang, Y. He, Y. Guo, T. Zhang and Y. Zhang, Dalton Trans., 2015, 44, 4297–4307 RSC.
- H. Katsumata, T. Sakai, T. Suzuki and S. Kaneco, Ind. Eng. Chem. Res., 2014, 53, 8018–8025 CrossRef CAS.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- M. Miyauchi, Y. Nukui, D. Atarashi and E. Sakai, ACS Appl. Mater. Interfaces, 2013, 5, 9770–9776 CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- B. Lei, M. Zhu, P. Chen, C. Chen, W. Ma, T. Li and M. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 4160–4169 CAS.
- N. L. Hawari and M. R. Johan, J. Alloys Compd., 2011, 509, 2001–2006 CrossRef CAS.
- Z.-M. Yang, G.-F. Huang, W.-Q. Huang, J.-M. Wei, X.-G. Yan, Y.-Y. Liu, C. Jiao, Z. Wan and A. Pan, J. Mater. Chem. A, 2014, 2, 1750–1756 CAS.
- W. Jiang, C. An, J. Liu, S. Wang, L. Zhao, W. Guo and J. Liuc, Dalton Trans., 2014, 43, 300–305 RSC.
- H. Li, G. Wang, F. Zhang, Y. Cai, Y. Wang and I. Djerdj, RSC Adv., 2012, 2, 12413–12423 RSC.
- S. Tsunekawa, T. Fukuda and A. Kasuya, Surf. Sci., 2000, 457, L437–L440 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari, S. Kalathil, J. Lee and M. H. Cho, RSC Adv., 2014, 4, 16782–16791 RSC.
- L. T. Murciano, A. Gilbank, B. Puertolas, T. Garcia, B. Solsona and D. Chadwick, Appl. Catal., B, 2013, 132–133, 116–122 CrossRef.
- D. A. Reddy, C. Liu, R. P. Vijayalakshmi and B. K. Reddy, Ceram. Int., 2014, 40, 1279–1288 CrossRef CAS.
- H. R. Safaei, M. Safaeib and M. Shekouhy, RSC Adv., 2015, 5, 6797–6806 RSC.
- R. S. Das, B. Singh, R. Banerjee and S. Mukhopadhyay, Dalton Trans., 2013, 42, 4068–4080 RSC.
- N. Soltani, E. Saion, M. Erfani, K. Rezaee, G. Bahmanrokh, G. P. C. Drummen, A. Bahrami and M. Z. Hussein, Int. J. Mol. Sci., 2012, 13, 12412–12427 CrossRef CAS PubMed.
- D. A. Reddy, D. H. Kim, S. J. Rhee, C. U. Jung, B. W. Lee and C. Liu, J. Alloys Compd., 2014, 588, 596–604 CrossRef.
- D. A. Reddy, G. Murali, B. Poornaprakash, R. P. Vijayalakshmi and B. K. Reddy, Appl. Surf. Sci., 2012, 258, 5206–5211 CrossRef CAS.
- R. Vinoth, P. Karthik, C. Muthamizhchelvan, B. Neppolian and M. Ashokkumar, Phys. Chem. Chem. Phys., 2016, 18, 5179–5191 RSC.
- Y.-W. Zhang, R. Si, C.-S. Liao, C.-H. Yan, C.-X. Xiao and Y. Kou, J. Phys. Chem. B, 2003, 107, 10159–10167 CrossRef CAS.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS.
- N. Wetchakun, S. Chaiwichain, B. Inceesungvorn, K. Pingmuang, S. Phanichphant, A. I. Minett and J. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 3718–3723 CAS.
- H. Cheng, B. Huang, Y. Dai, X. Qin and X. Zhang, Langmuir, 2010, 26, 6618–6624 CrossRef CAS PubMed.
- S. Kumar, J. Mater. Sci.: Mater. Electron., 2011, 22, 244–247 CrossRef CAS.
- H. Huang, D. Li, Q. Lin, W. Zhang, Y. Shao, Y. Chen, M. Sun and X. Fu, Environ. Sci. Technol., 2009, 43, 4164–4168 CrossRef CAS PubMed.
- N. Shehata, K. Meehan, M. Hudait and N. Jain, J. Nanopart. Res., 2012, 14, 1173–1183 CrossRef.
- L. Li, H. Wang, L. Zou and X. Wang, RSC Adv., 2015, 5, 41506–41512 RSC.
- S. Maensiri, C. Masingboon, P. Laokul, W. Jareonboon, V. Promarak, P. L. Anderson and S. Seraphin, Cryst. Growth Des., 2007, 7, 950–955 CAS.
- J. Li, O. Zalloum, T. Roschuk, C. Heng, J. Wojcik and P. Mascher, Appl. Phys. Lett., 2009, 94, 011112 CrossRef.
- F. Meng, L. Wang and J. Cui, J. Alloys Compd., 2013, 556, 102–108 CrossRef CAS.
- S. Phokha, S. Pinitsoontorn, P. Chirawatkul, Y. Poo-arporn and S. Maensiri, Nanoscale Res. Lett., 2012, 7, 1–13 CrossRef PubMed.
- C. Li, R. Hu, T. Zhou, H. Wu, K. Song, X. Liu and R. Wang, Mater. Lett., 2014, 124, 81–84 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and M. Karimi-Shamsabadi, Chem. Eng. J., 2013, 228, 631–641 CrossRef CAS.
- S. Zhuang, X. Xu, B. Feng, J. Hu, Y. Pang, G. Zhou, L. Tong and Y. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 613–621 CAS.
- D. A. Reddy, J. Choi, S. Lee, R. Ma and T. K. Kim, RSC Adv., 2015, 5, 67394–67404 RSC.
- Z. Zhang, W. Wang, L. Wang and S. Sun, ACS Appl. Mater. Interfaces, 2012, 4, 593–597 CAS.
- X. Zhou, J. Lan, G. Liu, K. Deng, Y. Yang, G. Nie, J. Yu and L. Zhi, J. Angew. Chem., Int. Ed., 2012, 51, 178–182 CrossRef CAS PubMed.
- S. Gu, Y. Chen, X. Yuan, H. Wang, X. Chen, Y. Liu, Q. Jiang, Z. Wuab and G. Zengab, RSC Adv., 2015, 5, 79556–79564 RSC.
- Q. Wang, S. Yu, Z. Tan, R. Zhang, Z. Li, X. Gao, B. Shen and H. Su, CrystEngComm, 2015, 17, 671–677 RSC.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS PubMed.
- G. Li, K. H. Wong, X. Zhang, C. Hu, J. C. Yu, R. C. Y. Chan and P. K. Wong, Chemosphere, 2009, 76, 1185–1191 CrossRef CAS PubMed.
- M. Balestrieri, S. Colis, M. Gallart, G. Schmerber, M. Ziegler, P. Gilliot and A. Dinia, J. Mater. Chem. C, 2015, 3, 7014–7021 RSC.
- S. Askrabic, Z. D. Dohcevic-Mitrovic, V. D. Araujo, G. Ionita, M. M. de Lima Jr and A. Cantarero, J. Phys. D: Appl. Phys., 2013, 46, 495306 CrossRef.
- L. Ye, K. Deng, F. Xu, L. Tian, T. Peng and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 82–85 RSC.
- W. Li, D. Li, Y. Lin, P. Wang, W. Chen, X. Fu and Y. Shao, J. Phys. Chem. C, 2012, 116, 3552–3560 CAS.
- Y. Lin, D. Li, J. Hu, G. Xiao, J. Wang, W. Li and X. Fu, J. Phys. Chem. C, 2012, 116, 5764–5772 CAS.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- P. Trogadas, J. Parrondo and V. Ramani, ACS Appl. Mater. Interfaces, 2012, 4, 5098–5102 CAS.
- Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wand and M. Fan, RSC Adv., 2014, 4, 13610–13619 RSC.
- C. Shifu, J. Lei, T. Wenming and F. Xianliang, Dalton Trans., 2013, 42, 10759–10768 RSC.
- J. Weimmerskirch-Aubatin, M. Stoffel, X. Devaux, A. Bouché, M. Vergnat and H. Rinnert, Phys. Status Solidi C, 2014, 11, 1630–1633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; EDS element mapping; EDS spectrum of AgI, CA-2 nanocomposite and CeO2; XPS spectrum of AgI and CeO2; UV-Vis absorption spectra of RhB aqueous solution containing CeO2, AgI, and CeO2–AgI (10, 20, 30, 40 wt%, respectively); digital image of the aliquots of RhB dye solutions in the presence of AgI and CA-2 nanocomposite; absorption spectra of phenol solution in the presence of CA-2 and AgI photocatalysts, and comparison for the degradation of phenol; XRD patterns of the 20 wt% CeO2–AgI nanocomposite after 5 recycling runs. See DOI: 10.1039/c5ra27533d |
| This journal is © The Royal Society of Chemistry 2016 |