
Formation of cobalt silicide nanoparticles on graphene with a synergistic effect and high stability for ethanol oxidation
Mingmei Zhang*,
Denghui Pan,
Yuan Li,
Zaoxue Yan,
Suci Meng and
Jimin Xie
School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: jdzmm@163.com; Fax: +86 11 88791800; Tel: +86 11 88791708
First published on 7th March 2016
Abstract
Stable cobalt silicide (CoSi) with an average diameter of less than 4 nm is uniformly decorated with graphene by a chemical vapor deposition method. Palladium (Pd) is then supported on the CoSi–G composite (Pd/CoSi–G) using an intermittent microwave heating reduction method and this composite is used as an electrocatalyst for ethanol oxidation in alkaline media. The Pd/CoSi–G catalyst exhibits 1.8 and 4.3 times the peak current density compared with Pd loaded on graphene and Vulcan XC-72 carbon, respectively. The significant increase in catalytic activity can be contributed to CoSi benefitting the formation of smaller and highly dispersed Pd particles and the synergistic effect between CoSi and Pd. The novel multi-effects of Pd on CoSi make Pd/CoSi–G a highly active, durable and low-cost fuel cell electrocatalyst candidate.
1. Introduction
In recent years, Pd-based electrocatalysts have drawn much attention because they are very suitable as an alternative material to Pt due to the low cost of Pd compared with Pt and they desirably overcome the CO-poisoning effect for alcohol oxidation in alkaline media.1–3 The last few decades have witnessed the emergence of various Pd-based nanomaterials and their wide applications.4–6 For instance, a great deal of effort has been devoted to reducing the use of Pd and enhancing catalytic efficiency. Zhang et al.7 reported that a core–shell catalyst composed of Ni and Pd obviously reduced palladium loading and had a higher catalytic activity toward ethanol oxidation than Pd metal alone. Lv and co-workers synthesized Pd–Pt nanodendrites with enhanced electrocatalytic activity for methanol oxidation, compared with commercial Pd black and Pt black catalysts.8 Researchers have pointed out that the electric ligand effect and downshift in d-band energy center may together promote the electrochemical activity of the alloyed electrocatalysts.9–11 Another way to increase the catalytic activity of the Pd-based catalyst is by using a suitable catalyst promoter. Yan et al. found that the electrocatalytic activity of Pd supported on MoC for ethanol oxidation in alkaline media was 3 times as high as that of Pd supported on carbon.12 Transition metal carbides are effective in promoting noble metal based electrocatalysis because of their synergistic effect on noble metals. This synergistic effect is caused by electron transfer between the carbides and noble metals.13 Despite this being demonstrated, it is still of practical importance to develop novel Pd-based catalysts with enhanced catalytic performance for progress in fuel cells. Since transition metal silicides (MSi, M = Co, Fe, Ni) have Pt-like catalytic properties and could be used instead of some noble metal catalysts in catalyzing aromatic hydrogenation and naphthalene hydrogenation,14–18 they are more and more popular and important in the field of catalysis. However, transition metal silicides have large particle sizes and low surface area, which restricts their application in catalysis. It is an important challenge to synthesize small and well-dispersed metal silicides for carbon supports under controllable conditions. Recently, our research team was involved in the development of new synthesis routes for metal silicides to obtain new low-cost electrocatalysts. In summary, highly dispersed CoSi nanoparticles supported on graphene were successfully obtained. The preparation of smaller CoSi nanoparticles below 5 nm in diameter is critical in order to enhance the synergistic effect of CoSi nanoparticles with noble metal nanoparticles. In addition, Yan et al. reported an intermittent microwave heating method to reduce the sizes of noble metal particles.19 This method could let Pd nuclei grow intermittently and avoid big particles, which enhances the catalytic activity by a large degree.Here, we report a metal organic chemical vapor deposition method of producing CoSi nanoparticles with controllable sizes ranging from 1 to 3 nm on a graphene surface. To the best of our knowledge, this is the first attempt at preparing Pd nanoparticles supported on such small CoSi nanoparticles using an intermittent microwave heating method. The Pd/CoSi–G electrocatalyst showed highly active and stable catalytic performance compared with that of Pd/G and commercial Pd/C. The increased catalytic activity and stability were respectively ascribed to the synergistic effect and the strong interaction forces between Pd and CoSi. The results show Pd/CoSi–G to be a promising candidate as a fuel cell electrocatalyst.
2. Experimental
2.1. Materials
Natural flake graphite was obtained from Qingdao Guyu graphite Co., Ltd. with a particle size of 150 nm. Ethylene glycol (EG), trichlorosilane (SiHCl3), cobalt carbonyl (Co2(CO)8) and chloropalladium acid (H2PdCl4) were purchased from Sinopharm Chemical Reagent Co., Ltd., China and used as received without any further purification. Distilled water was also used throughout the experiments.2.2. Preparation of CoSi–graphene composites
Typically, a Co(SiCl3)(CO)4 precursor was synthesized under an atmosphere of dry argon using Schlenk line techniques according to the literature.20,21 20 mL of freshly distilled SiHCl3 was added to 2.0 g of Co2(CO)8 placed in a Carius tube, cooled to −40 °C in an acetonitrile–dry ice slurry and stirred for 2.5 h before warming to room temperature. Excess trichlorosilane was removed in vacuo. The residual grey-brown powder was purified by repeated sublimation at 45 °C and 0.1 mbar. A final product was obtained as yellow crystals. GO was prepared from purified natural graphite using a modified method from our previous work; 3.0 g of graphite powder was concentrated with H2SO4 (150 mL) and NaNO3 (6.0 g) solution in a 500 mL flask. With vigorous stirring, KMnO4 (15.0 g) was added gradually and the temperature of the mixture was kept at below 10 °C. The suspension was removed from the ice bath and the temperature was allowed to reach 80 °C for 2 h until it became pasty brownish, and then diluted with distilled water (100 mL). Following this, the mixture was stirred for 30 min and 50 mL of 30 wt% H2O2 was added. The mixture was filtered and washed with 5% HCl aqueous solution to remove metal ions followed by distilled water to remove the acid. For further purification, the as-obtained graphite oxide was re-dispersed in distilled water and then was dialyzed for one week to remove residual salts and acids. The final product was dried under vacuum and stored in vacuum desiccators until use. The Co(SiCl3)(CO)4 precursor was sublimed and adsorbed on the GO support at three concentrations of 40, 30 and 20 wt%, and equally divided into 3 parts at 50 °C under vacuum. Then, the GO adsorbed precursor was put in a quartz-tube heater with a flow rate of 30 mL min−1 argon for 2 h at room temperature to remove oxygen in the reactor and keep the reaction under an inert atmosphere. The three GO adsorbed precursors were treated under 200 mL min−1 of 10% H2/Ar at 300 °C for 120 min at atmospheric pressure, and were denoted as CoSi–G-4, CoSi–G-3 and CoSi–G-2. The corresponding average CoSi nanoparticle sizes were determined to be 5.7 nm, 2.5 nm and 2.0 nm, respectively.2.3. Preparation of the Pd/CoSi–graphene electrocatalyst
Pd supported on CoSi–G-3 (denoted as Pd/CoSi–G-3) or graphene (denoted as Pd/G) was prepared and used as an electrocatalyst for ethanol oxidation. Typically, H2PdCl4 in 50 mL of ethylene glycol (containing 40 mg of Pd) was mixed with 60 mg of CoSi–G-3 or graphene, treated in an ultrasonic bath for 30 min and vigorously stirred for 24 h at room temperature to form a uniform ink. The pH of the mixture was adjusted to 9.5 with 2.0 M NaOH/glycol solution. The sample was then put into a microwave oven (800 W) for intermittent microwave heating using a 15 s on and 10 s off procedure repeated 10 times. Ethylene glycol as the reducing agent made Pd(II) into Pd(0) and oxalic acid was formed during the reaction. After cooling to room temperature, the product was washed with distilled water 5–6 times to remove excess ethylene glycol and subsequently separated by sintered discs and dried under vacuum at 80 °C for 10 h. The theoretical Pd content in Pd/CoSi–G-3 targeted was 40 wt%. Inductively coupled plasma spectroscopy (ICP, IRIS(HR), USA) analysis gave the actual Pd content to be 38.9 wt%.2.4. Preparation of the catalyst electrode
For the electrode preparation, Pd/CoSi–G-3 or Pd/G (10 mg) was dispersed in 1 mL of ethanol and 0.5 mL of 0.5 wt% Nafion suspension (DuPont, USA) under ultrasonic agitation to form an electrocatalyst ink. The electrocatalyst ink (10 μL) was then deposited on the surface of a glassy carbon rod and dried at room temperature overnight. Total Pd loading was controlled at 0.05 mg cm−2. All chemicals were of analytical grade and used as received.2.5. Characterization of the supports and electrocatalysts
The morphology of the as-prepared samples was examined using a TEM (JEOL-JEM-2010, Japan) operated at 120 kV. X-ray photoelectron spectroscopy (XPS) was recorded on a PHI-5702, and the C 1s line at 291.4 eV was used as the binding energy reference. The structures of the obtained samples were examined by XRD using D8 Advance X-ray powder diffraction (Bruker AXS company, Germany) equipped with Cu K R radiation (λ) 1.5406 (Å), employing a scanning rate of 0.02° s−1 in a 2θ range from 10° to 80°. The graphitization degree of these samples was determined by Laser Micro-Raman Spectrometer (Renishaw inVia, Renishaw plc, UK). Electrochemical measurements were performed by cyclic voltammetry (CV) using an IM6e potentiostat (Zahner-Electrik, Germany) at 30 °C controlled by a water bath thermostat. The three-electrode cell system consisted of a pre-treated glassy carbon working electrode (0.05 cm2) and platinum foil (1.0 cm2) and Hg/HgO electrodes used as counter and reference electrodes, respectively.3. Results and discussion
A schematic illustration of the formation of the Pd/CoSi–G nanocomposite is shown in Fig. 1. Once graphite oxide was synthesized, the Co(SiCl3)(CO)4 precursor was sublimed and adsorbed on the GO support at 50 °C under vacuum. Then, the GO adsorbed precursor was treated in a vacuum tube furnace under atmospheric pressure, which led to a stable black CoSi–graphene. Finally the CoSi–graphene was added to a H2PdCl4 solution and put into a microwave oven for intermittent microwave heating; well dispersed Pd nanoparticles with small sizes on CoSi–graphene were obtained. | ||
| Fig. 1 Schematic diagram of the synthesis of the Pd/CoSi–G electrocatalyst. | ||
Fig. 2 shows the TEM images of the CoSi on graphene surface. Fig. 2a shows the TEM image of the CoSi–G-4 sample synthesized at a higher content of Co(SiCl3)(CO)4 precursor. The distribution of the CoSi–G-4 nanoparticles is not very uniform and the average particle size is larger than 5.7 nm in diameter (Fig. 1b). Fig. 2c indicates that the sample prepared at medium content of Co(SiCl3)(CO)4 precursor yields much smaller CoSi size with better dispersion and distribution (CoSi–G-3). The mean size of the CoSi nanoparticles on graphene obtained by measuring 100 randomly chosen particles in the magnified TEM images was 2.5 nm (Fig. 2d). Smaller CoSi particles could be prepared when a lower content of Co(SiCl3)(CO)4 precursor adsorbed on GO was used in the synthesis. Typically, we prepared CoSi nanoparticles with an average size of 2.0 nm (CoSi–G-2), as shown in Fig. 2e and f. The results confirm that CoSi nanoparticles are successfully synthesized on the graphene surface by a chemical vapor deposition method under such experimental conditions.
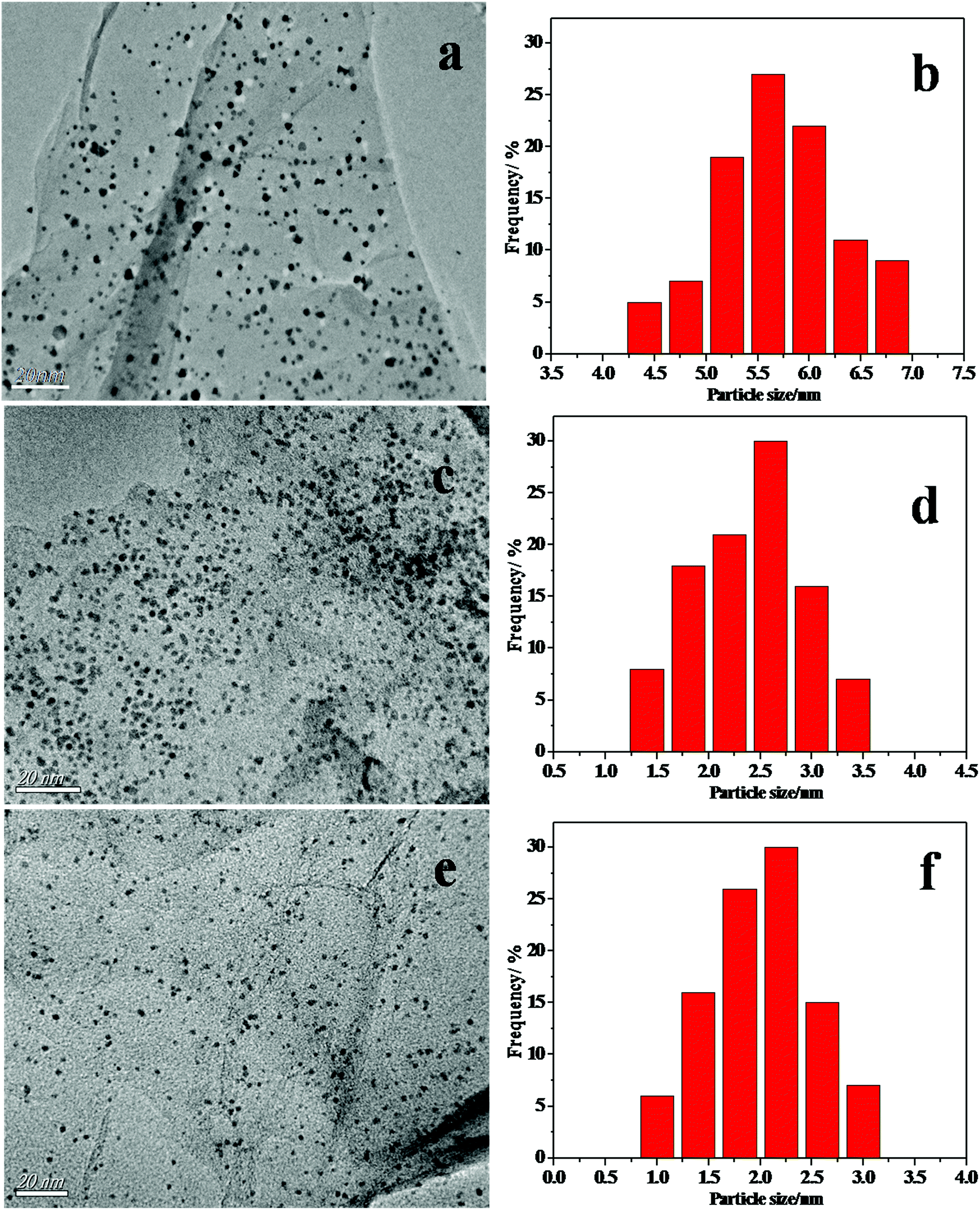 | ||
| Fig. 2 The TEM images and histograms of the materials (a) CoSi–G-4, (c) CoSi–G-3 and (e) CoSi–G-2. The CoSi particle size distributions of (b) CoSi–G-4, (d) CoSi–G-3 and (f) CoSi–G-2. | ||
Fig. 3 shows the XRD patterns of Pd/CoSi–G-3 (a), CoSi–G-3 (b) and GO (c). All the patterns match the characteristics of CoSi (PDF#50-1337) by comparing the JCPDS card. The diffraction peaks in Fig. 2b at 2θ = 40.53°, 45.97°, 50.36° and 80.95° correspond to the (200), (210), (211) and (321) facets of the CoSi crystal, respectively. The results also exhibit that the CoSi crystal can be successfully synthesized by metal organic chemical vapor deposition of Co(SiCl3)(CO)4 as a precursor supported on graphene. The peaks in Fig. 3a at 2θ = 40.1°, 46.9° and 68.1° correspond to the (111), (200) and (220) facets of the Pd crystal, respectively. The CoSi (210) peak and the Pd (220) peak can be used to calculate the crystal size according to Scherrer’s equation. The calculated mean sizes of the Pd and CoSi particles of the Pd/CoSi–G-3 electrocatalyst were found to be 2.2 nm and 2.5 nm, respectively.
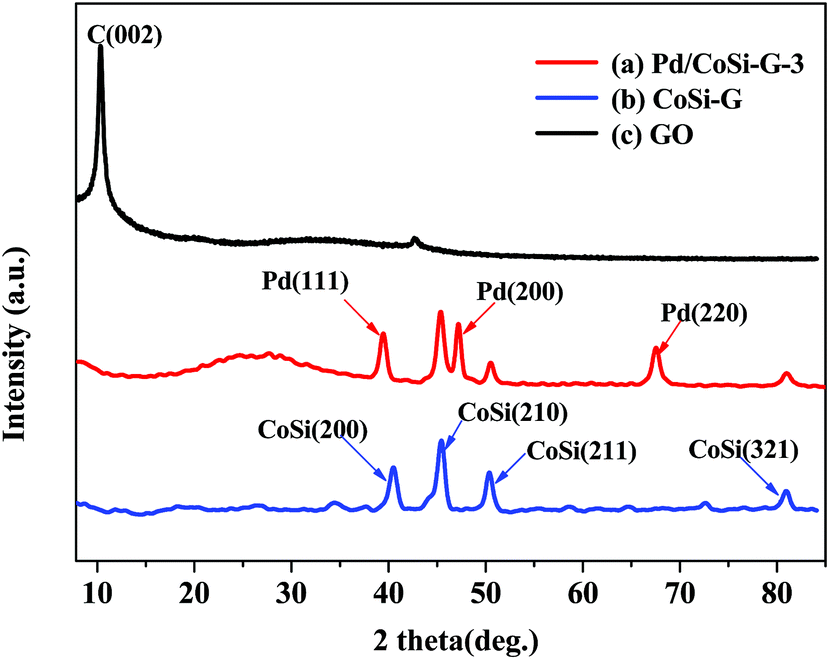 | ||
| Fig. 3 XRD patterns of (a) Pd/CoSi–G-3, (b) CoSi–G-3 and (c) GO. | ||
The crystalline nature and particle size of the Pd/CoSi–G-3 samples were confirmed by transmission electron microscopy (TEM) measurements. The TEM image in Fig. 4a shows that uniform nanostructured CoSi particles are evenly dispersed on the graphene surface. The diameters of CoSi nanoparticles range from 1.5 nm to 3.5 nm and the mean diameter was about 2.5 nm, which is in good agreement with the XRD results. Fig. 4b shows that a large number of Pd nanoparticles occupy the majority of the surface of CoSi–G-3 sheets with a fairly even, dense distribution, indicating a strong interaction between the Pd nanoparticles and the CoSi supported graphene sheets. A high resolution transmission electron microscopy (HRTEM) image (Fig. 4c) clearly shows visible crystal lattice fringes of CoSi (210) and Pd (111), further confirming the coexistence of CoSi and Pd. The energy dispersive spectroscopy (EDS) pattern of Pd/CoSi–G-3 in Fig. 4d proves the coexistence of Pd, Co, Si and C elements. The peaks of Cu elements come from the sample bracket. This further illustrates that the presence of CoSi on the graphene sheets could help the formation of small, highly concentrated, and uniformly dispersed Pd nanoparticles. Meanwhile, the very small CoSi particles and graphene could give an effective synergistic effect and improved mass transfer properties when used as a catalyst support. Pd nanoparticles were therefore loaded on the synthesized CoSi–G-3 materials and used for ethanol oxidation.
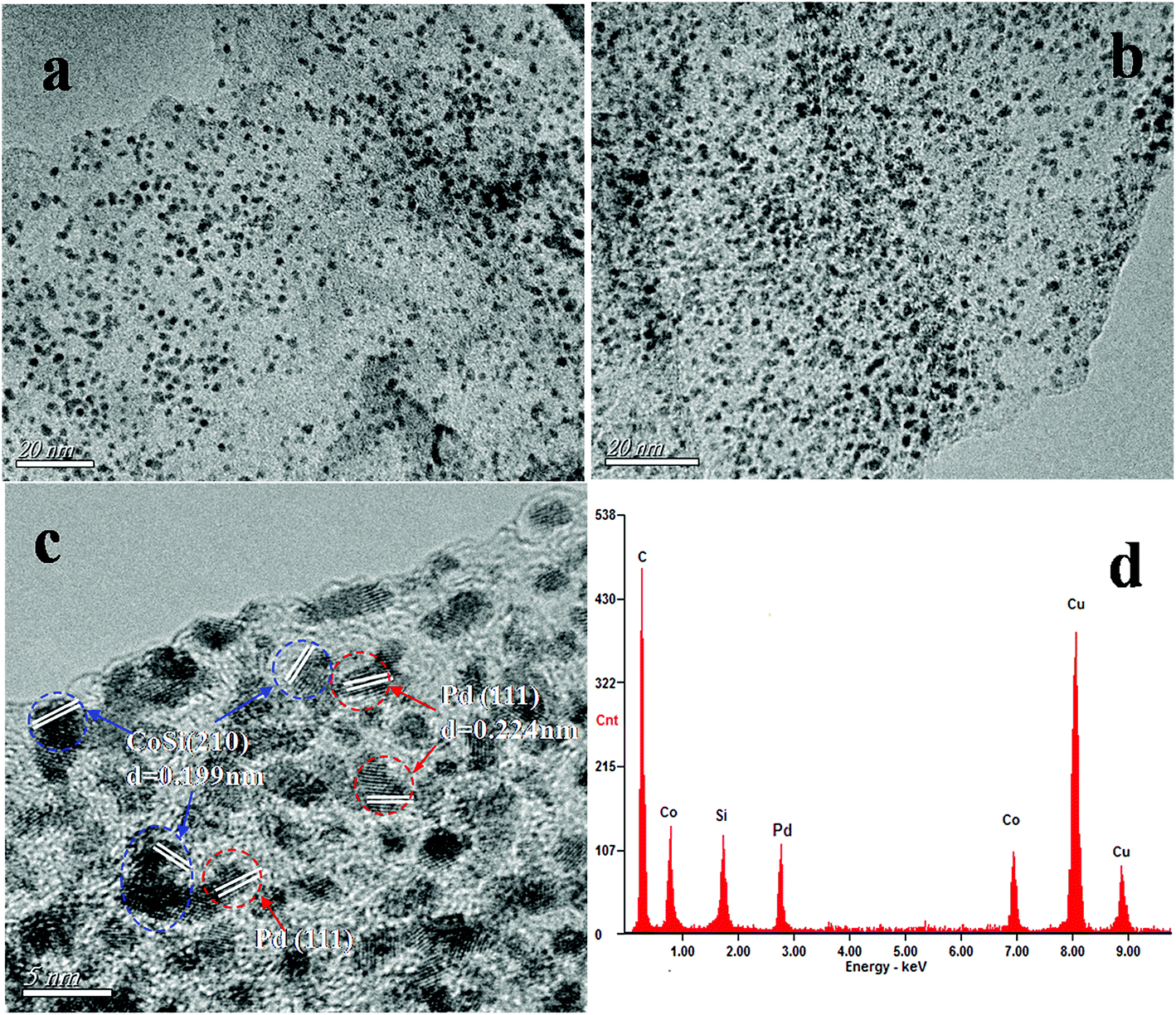 | ||
| Fig. 4 TEM images of (a) CoSi–G-3 and (b) Pd/CoSi–G-3, (c) HRTEM image of Pd/CoSi–G-3, and (d) EDS spectrum of the Pd/CoSi–G-3 composite. | ||
Raman spectroscopy is considered to be a very versatile optical method for the characterization of graphitic materials from their vibration spectra. The Raman spectra of GO, CoSi–G-3 and Pd/CoSi–G-3 are shown in Fig. 5. As can be seen, after the growth of CoSi–G-3 and Pd/CoSi–G-3, the D band (1348 cm−1) is the same, which means that the defects in the curved graphene sheet do not change obviously.22,23 However, the G band shifts to lower values for Pd/CoSi–G-3 (1582.02 cm−1) and CoSi–G-3 (1584.76 cm−1) compared with GO (1590.91 cm−1), which reveals the partial reduction of GO and restoration of the graphite structure. The integrated intensity ratio of the D and G bands (ID/IG) increases with the amount of disorder for graphitic materials, vanishing for completely defect-free graphite. As shown in Fig. 5 the ratios of the peak intensities of ID/IG for GO, CoSi–G-3 and Pd/CoSi–G-3 have been estimated to be around 0.99, 1.04 and 1.08, respectively. Such a higher ratio of the D peak to G peak in Pd/CoSi–G-3 (1.08) is attributable to the decrease in the average size of sp2 domains upon the formation of nanoparticles on the graphene and the reduction of exfoliated graphene.24,25
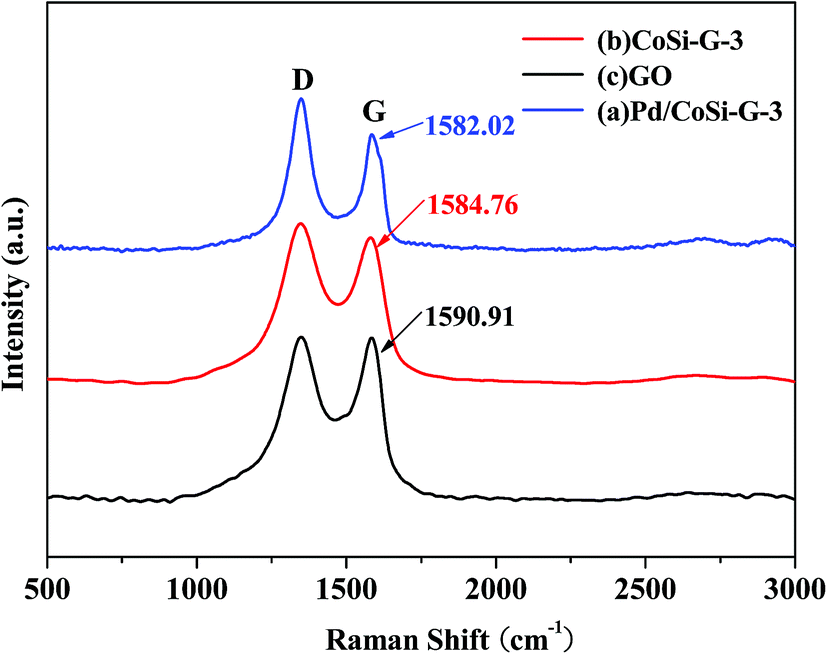 | ||
| Fig. 5 Raman spectra of Pd/CoSi–G-3 (a), CoSi–G-3 (b) and GO (c). | ||
The XPS spectra (Pd 3d) of Pd/G and Pd/CoSi–G-3 are presented in Fig. 6, which can be used to explain the electrocatalyst performance. The peaks of Pd/G at 339.98 eV and 334.68 eV belong to metallic Pd, and the peaks at 342.14 eV and 336.85 eV belong to Pd(II).26 However, Pd/CoSi–G-3 has peaks at 340.57 eV and 335.27 eV for metallic Pd, and 342.80 eV and 337.47 eV for Pd(II). Obviously, a positive shift occurs in the XPS spectra after the introduction of CoSi. This result indicates that there is increased electron-donating transfer between the Pd and CoSi compared with that between Pd and graphene, which might result in the less overpotential and higher mass activity of Pd/CoSi–G-3. On the other hand, the greater degree of electron transfer means a higher linkage between Pd and CoSi, which may account for the excellent electrocatalytic stability of Pd/CoSi–G-3.
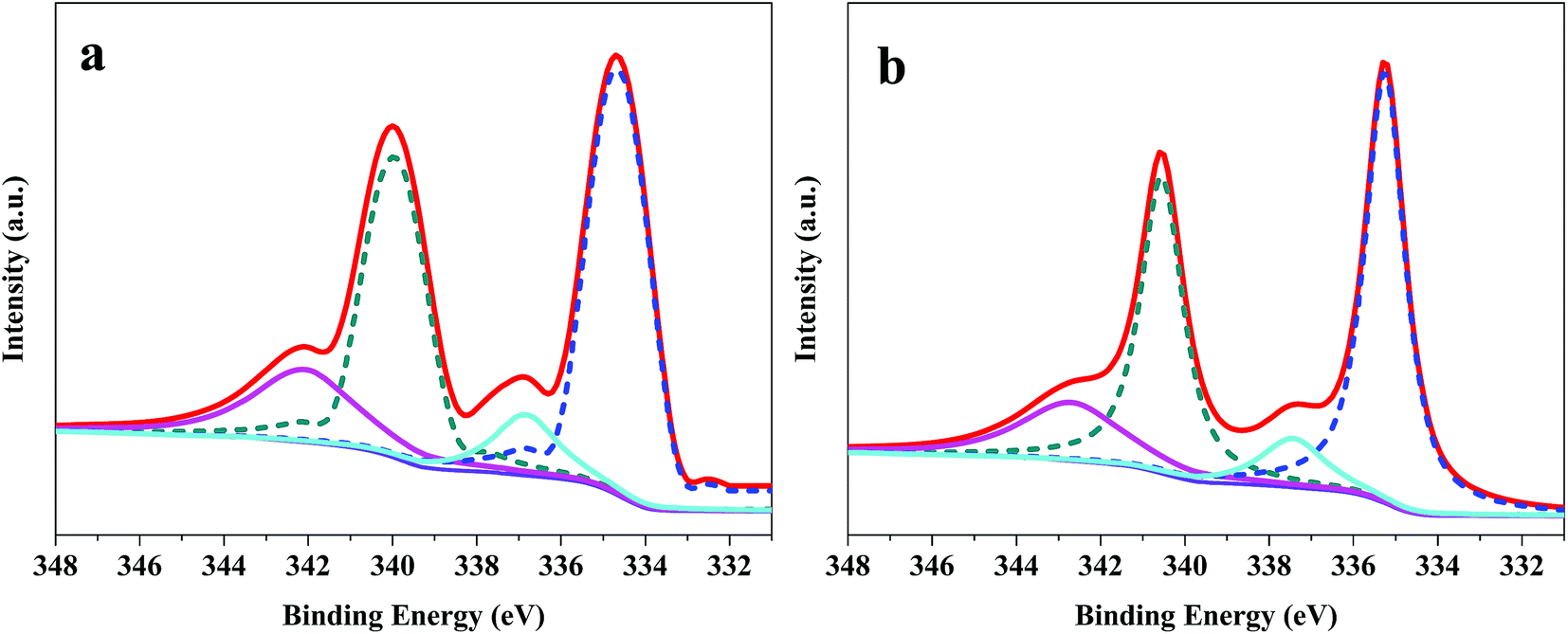 | ||
| Fig. 6 XPS spectra (Pd 3d) of (a) Pd/G and (b) Pd/CoSi–G-3. | ||
Fig. 7a shows the cyclic voltammograms (CVs) of alcohol electrooxidation on Pd/CoSi–G-3, Pd/G and Pd/C electrodes in 1.0 M KOH + 1.0 M ethanol solution at 303 K and at a scan rate of 50 mV s−1. The anodic peak current density (forward scan) reflects the amount of ethanol oxidized by the Pd electrocatalysts, and the anodic peak potential represents the ability of the electrocatalysts to catalyze ethanol oxidation. Therefore, the anodic peak current density and the peak potential are two important parameters to evaluate the electrocatalytic activity of the catalysts.27 For ethanol oxidation, the mass activity of ethanol oxidation in 1 mol dm−3 ethanol solution on Pd/CoSi–G-3 was 80.95 mA cm−2, which is higher than Pd/G (45.68 mA cm−2), and much higher than that of 18.75 mA cm−2 on a Pd/C electrode. It shows a 4.3 times higher peak current density than that of the Pd/C electrocatalyst at the same Pd loading under similar electrochemical reaction conditions. The results indicate that the great synergistic effect of CoSi on Pd leads to the significantly improved activity and output of Pd/CoSi–G-3 when used in fuel cells. The synergistic effect of CoSi on Pd can be further confirmed by comparing the peak current density and ECSA between Pd/CoSi–G-3, Pd/G and Pd/C electrodes (Fig. 7b). The electrochemical surface areas (ECSAs) of the Pd/CoSi–G-3, Pd/G and Pd/C electrodes catalysts were studied by CV tests from −0.70 to 0.20 V in 1.0 mol L−1 KOH at a scan rate of 50 mV s−1, and were calculated based on the PdO reduction peak changing, and the assumption of 212 μC cm−2 of the electrode surface.28 They are 157.4 m2 g−1, 98.37 m2 g−1and 44.97 m2 g−1 for Pd/CoSi–G-3, Pd/G and Pd/C, respectively. The ECSA on Pd/CoSi–G-3 is 1.6 times higher than that of Pd/G, confirming the great synergistic effect of MoC on Pd. The activities of the Pd/CoSi–G electrocatalyst were compared to others in the literature. We found that the electrochemical surface areas (ECSAs) of the Pd/CoSi–G are 1.3–6.5 times higher than the reported Pd/graphene electrocatalysts,29,30 Pd–Ag alloy supported electrocatalysts,31 and Pd supported carbon electrocatalysts under similar electrochemical reaction conditions.32 Moreover, the high surface area of CoSi–G-3 as a support is beneficial for the uniform dispersion of the Pd nanoparticles to make them highly used for easier mass transfer, which display most commercial competition. As for detecting the mechanism of ethanol oxidation on CoSi modified Pd electrocatalyst, more efforts will be needed.
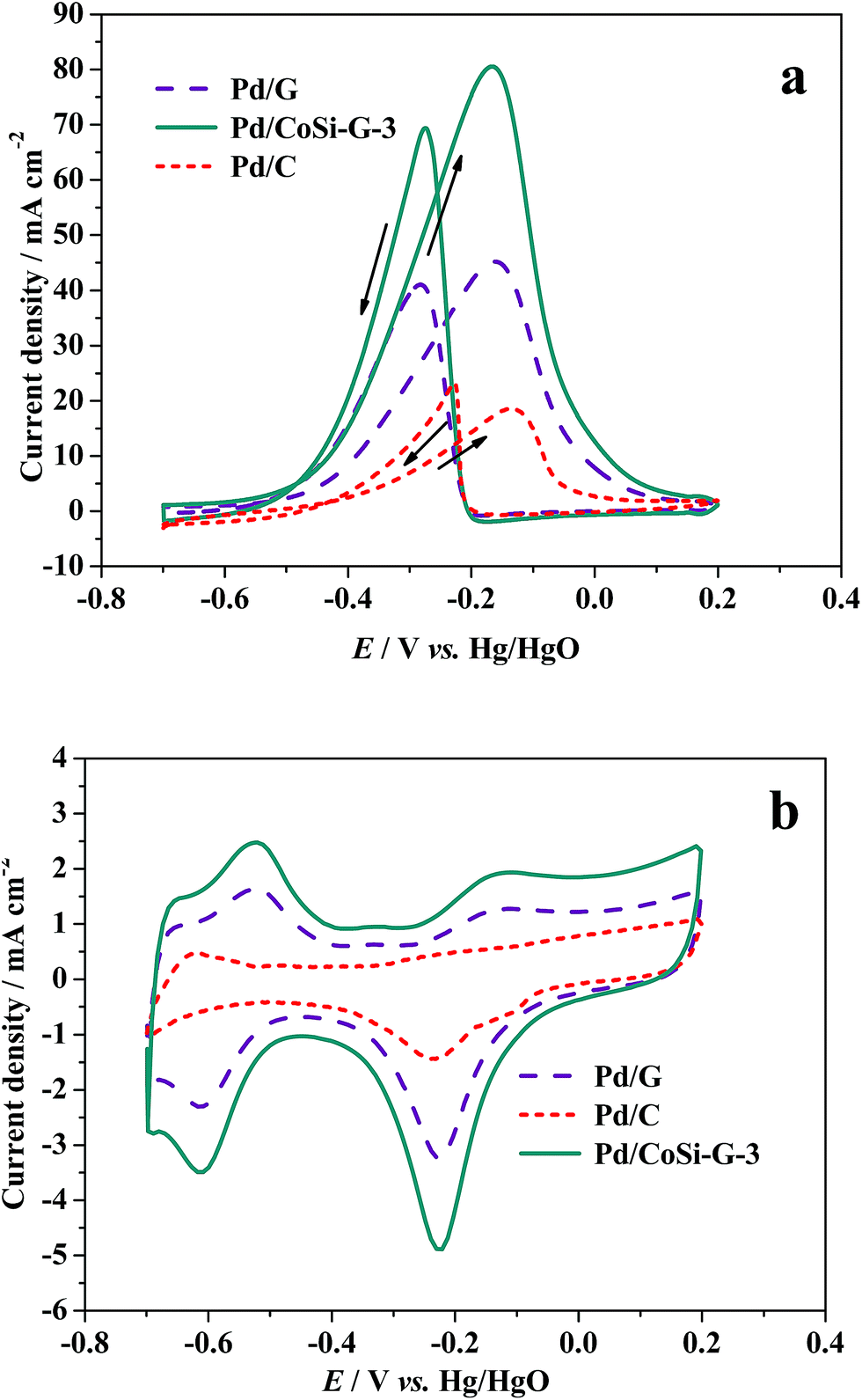 | ||
| Fig. 7 (a) Cyclic voltammograms of ethanol oxidation on Pd/CoSi–G-3, Pd/G and Pd/C electrodes in 1.0 M KOH/1.0 M ethanol solution at 303 K, scan rate: 50 mV s−1, and (b) cyclic voltammograms of the above electrodes in 1.0 M KOH solution at 303 K, scan rate: 50 mV s−1. | ||
In addition, the ratio of the forward anodic peak current density (If) to the reverse anodic peak current density (Ib), If/Ib, can be used to describe the catalyst tolerance to carbonaceous species accumulation. A higher ratio indicates more effective removal of the poisoning species on the catalyst surface during the anodic scan and little accumulation of carbonaceous residues on the electrode surface.33,34 The If/Ib ratio for the Pd/CoSi–G-3 catalyst was 1.17, which is higher than that of Pd/G (If/Ib = 1.08) indicating that more intermediate carbonaceous species were oxidized to carbon dioxide in the forward scan on the CoSi–G-3 surface than that on the surface of graphene.
The stability of a catalyst is of great importance for its commercial applications in fuel cells.35 The stability of Pd/CoSi–G-3 electrocatalyst for ethanol oxidation was also studied and is shown in Fig. 8. The shadows in Fig. 8 are the cycling results between the 1st cycle and the 4000th cycle. It is clear that the peak current density of the Pd/CoSi–G-3 reduced from 80.95 mA cm−2 to 73.94 mA cm−2, a reduction of 8.6%. However, the peak current density of Pd/G reduced by 21.03% from 45.46 mA cm−2 to 35.9 mA cm−2. Therefore, Pd/CoSi–G-3 has a greater catalytic stability than the Pd/G electrocatalyst. Meanwhile, reduction in the ECSA displays the same trend, as shown in Fig. 8b. The promoted electrochemical stability may be due to the stronger interaction forces between Pd and CoSi–graphene than the forces between Pd and G.
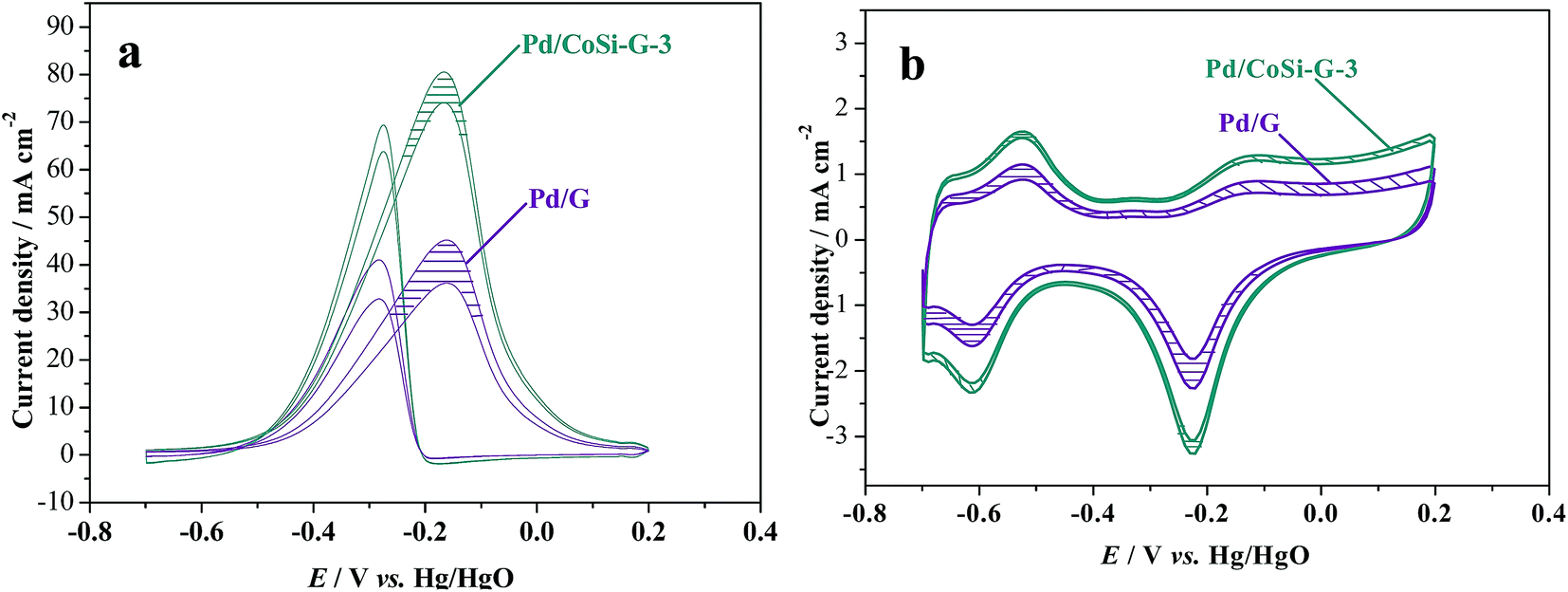 | ||
| Fig. 8 (a) Cyclic voltammograms of methanol oxidation on Pd/CoSi–G-3 and Pd/G electrodes in 1.0 mol L−1 ethanol/1.0 mol L−1 KOH solution with a scan rate of 50 mV s−1 and (b) the cyclic voltammograms on Pd/CoSi–G-3 and Pd/G electrodes in 1.0 mol L−1 KOH solution with a scan rate of 50 mV s−1. The shadows show the difference between the 1st cycle and the 4000th cycle. | ||
The chronopotentiometric testing further proves that the Pd/CoSi–G-3 electrocatalyst could sustain larger current densities for stable methanol oxidation than the Pd/G electrocatalyst as shown in Fig. 9. The Pd/CoSi–G-3 electrode could retain its catalytic performance for more than 1 h at a constant current density of 1.2 mA cm−2. When the constant current density increased up to 2.0 mA cm−2, the electrode performance was still sustained for more than 30 min. However, the Pd/G electrode could sustain just less than 50 min at the same current density and the same Pd content. A fast increase in the potential means a fast loss of activity. The results indicate that the Pd/CoSi–G-3 electrocatalyst could sustain larger current densities for stable ethanol oxidation than the Pd/G electrocatalyst.
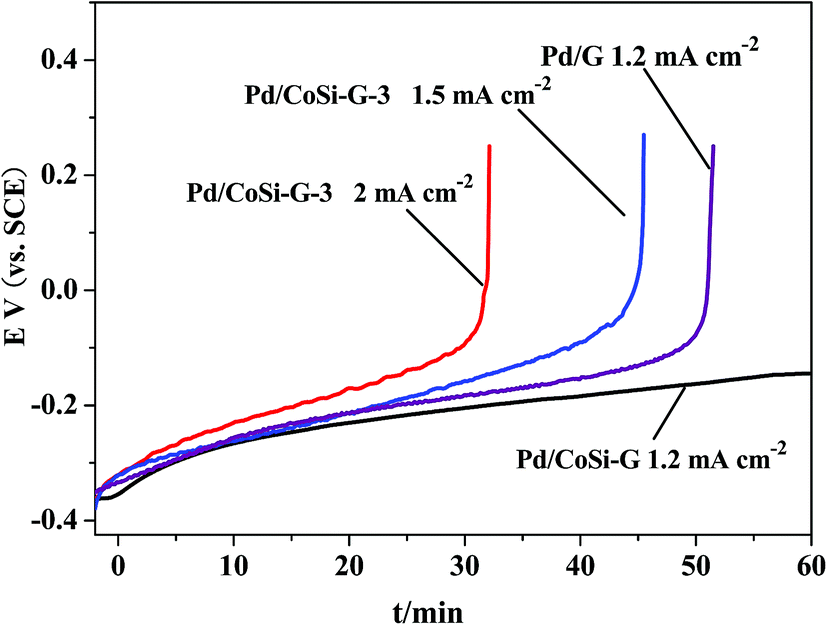 | ||
| Fig. 9 Chronopotentiometric curves of ethanol oxidation on Pd/CoSi–G-3 and Pd/G electrodes at different current densities in 1.0 mol L−1 ethanol/1.0 mol L−1 KOH solution. | ||
4. Conclusions
CoSi nanoparticles on graphene were successfully synthesized through metal organic chemical vapor deposition. The sizes of the CoSi particles from 1.5 nm to 3.5 nm can be easily controlled by adjusting the experimental parameters of this method. Pd nanoparticles were then loaded onto the CoSi–graphene to form a novel, stable structured Pd/CoSi–G-3 electrocatalyst, which showed much higher activity and more stable performance in the electrooxidation of ethanol than a Pd/G electrocatalyst and is even superior to Pd/C in alkaline media. The origin of the excellent performance of Pd/CoSi–G-3 should be mainly due to the inherent stability of CoSi and its electron-donating (synergistic effect) on Pd, which not only improves the activity by facilitating the oxidation of ethanol, but also improves the stability by increasing the linkage strength between Pd and CoSi. The outstanding electrocatalytic properties of Pd/CoSi–G-3 would make it a promising candidate for a variety of catalysis applications in fuel cells and others.Acknowledgements
This work was supported financially by the Natural Science Foundations of Jiangsu (BK20140531), Zhenjiang Industry Supporting Plan (GY2014040), China Postdoctoral Science Foundation (2015M570410) and Research Foundation for Talented Scholars of Jiangsu University (14JDG187); Dr Suci Meng thanks the National Natural Science Foundation of China (21103073).References
- R. N. Singh and R. Awasthia, Catal.: Sci. Technol., 2011, 1, 778 Search PubMed.
- J. N. Zheng, S. S. Li, X. Ma, F. Y. Chen, A. J. Wang, J. R. Chen and J. J. Feng, J. Power Sources, 2014, 262, 270 CrossRef CAS.
- Z. Yin, M. Chi, Q. Zhu, D. Ma, J. Sun and X. Bao, J. Mater. Chem. A, 2013, 1, 9157 CAS.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406 CrossRef CAS PubMed.
- S. Ghosh, H. Remita, P. Kar, S. Choudhury, S. Sardar, P. Beaunier, P. S. Roy, S. K. Bhattacharyae and S. K. Pala, J. Mater. Chem. A, 2015, 3, 9517 CAS.
- B. Li and J. Prakash, Electrochem. Commun., 2009, 11, 1162 CrossRef CAS.
- M. M. Zhang, Z. X. Yan, Q. Sun, J. M. Xie and J. J. Jing, New J. Chem., 2012, 36, 2533 RSC.
- J. J. Lv, J. N. Zheng, S. S. Li, L. L. Chen, A. J. Wang and J. J. Feng, J. Mater. Chem. A, 2014, 4384 CAS.
- Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122 CrossRef CAS.
- R. Awasthia and R. N. Singh, Catal.: Sci. Technol., 2012, 2, 2428 Search PubMed.
- D. Kim, Y. W. Lee, S. B. Lee and S. W. Han, Angew. Chem., Int. Ed., 2012, 51, 159 CrossRef CAS PubMed.
- Z. X. Yan, J. M. Xie, P. K. Shen, M. M. Zhang, Y. Zhang and M. Chen, Electrochim. Acta, 2013, 108, 644 CrossRef CAS.
- R. H. Wang, Y. Xie, K. Y. Shi, J. Q. Wang, C. G. Tian, P. K. Shen and H. G. Fu, Chem.–Eur. J., 2012, 18, 7443 CrossRef CAS PubMed.
- X. Chen, J. C. Guan, G. Y. Sha, Z. M. Gao, C. T. Williams and C. H. Liang, RSC Adv., 2014, 4, 653 RSC.
- C. H. Liang, V. Narkhede, J. ABmann and M. Muhler, Stud. Surf. Sci. Catal., 2006, 162, 473 CrossRef CAS.
- P. Serp, P. Kalck and R. Feurer, Chem. Rev., 2002, 102, 3085 CrossRef CAS PubMed.
- H. Z. Zhang, J. S. Qiu, C. H. Liang, Z. L. Li, X. Wang, Y. Wang, Z. C. Feng and C. Li, Catal. Lett., 2005, 101, 211 CrossRef CAS.
- C. H. Liang, A. Q. Zhao, X. F. Zhang, Z. Q. Ma and R. Prins, Chem. Commun., 2009, 2047 RSC.
- Z. X. Yan, M. M. Zhang, J. M. Xie, H. E. Wang and W. Wei, J. Power Sources, 2013, 243, 48 CrossRef CAS.
- M. M. Zhang, Z. X. Yan, Y. Li, J. J. Jing and J. M. Xie, Electrochim. Acta, 2015, 161, 48 CrossRef CAS.
- A. L. Schmitt, L. Zhu, D. Schmeiber, F. J. Himpsel and S. Jin, J. Phys. Chem. B, 2006, 110, 18142 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed.
- W. X. Wang, S. H. Liang, T. Yu, D. H. Li, Y. B. Li and X. F. Han, J. Appl. Phys., 2011, 109, 07C501 Search PubMed.
- P. Chen, T. Y. Xiao, H. H. Li, J. J. Yang, Z. Wang, H. B. Yao and S. H. Yu, ACS Nano, 2012, 6, 712 CrossRef CAS PubMed.
- T. Belin and F. Epron, Mater. Sci. Eng., B, 2005, 119, 105 CrossRef.
- J. J. Lv, J. N. Zheng, S. S. Li, L. L. Chen, A. J. Wang and J. J. Feng, J. Mater. Chem. A, 2014, 4384 CAS.
- S. An, J. H. Park, C. H. Shin, J. Joo, E. Ramasamy, J. Hwang and J. Lee, Carbon, 2011, 49, 1108 CrossRef CAS.
- Z. Y. Zhang, L. Xin, K. Sun and W. Z. Li, Int. J. Hydrogen Energy, 2011, 36, 12686 CrossRef CAS.
- Y. T. Zhang, H. H. Shu, G. Chang, K. Ji, M. Oyama, X. Liu and Y. B. He, Electrochim. Acta, 2013, 109, 570 CrossRef CAS.
- Y. Wang, Y. Zhao, W. T. He, J. Yin and Y. Q. Su, Thin Solid Films, 2013, 544, 88 CrossRef CAS.
- S. F. Fu, C. Z. Zhu, D. Du and Y. H. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 13842 CAS.
- E. Rikkinen, A. Santasalo-Aarnio, S. Airaksinen, M. Borghei, V. Viitanen, J. Sainio, E. I. Kauppinen, T. Kallio and A. O. I. Krause, J. Phys. Chem. C, 2011, 115, 23067 CAS.
- Y. H. Qin, H. H. Yang, X. S. Zhang, P. Li and C. A. Ma, Int. J. Hydrogen Energy, 2010, 35, 7667 CrossRef CAS.
- F. R. Nikkuni, E. A. Ticianelli, L. Dubau and M. Chatenet, Electrocatalysis, 2013, 4, 104 CrossRef CAS.
- L. Wang, C. Tian, B. Wang, R. Wang, W. Zhou and H. Fu, Chem. Commun., 2008, 5411 RSC.
| This journal is © The Royal Society of Chemistry 2016 |