
Oxyonium phosphobetaines – unusually stable nucleophilic catalyst–phosphate complexes formed from H-phosphonates and N-oxides†‡
Abstract
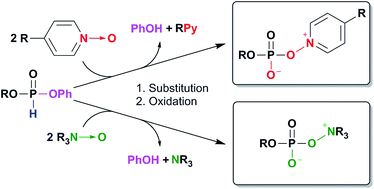
Aryl H-phosphonates react with N-oxides to form previously unknown stable zwitterionic oxyonium phosphates comprising an −O–P–O–N+Z atom system. Their structures were confirmed i.e. by X-ray crystal structure analysis, and some mechanisms were proposed for their formation. Stability during storage and reactivity toward nucleophiles points to their possible synthetic applications.
 Please wait while we load your content...
Please wait while we load your content...

