DOI:
10.1039/C5RA27397H
(Paper)
RSC Adv., 2016,
6, 15370-15381
Improved light absorbance does not lead to better DSC performance: studies on a ruthenium porphyrin–terpyridine conjugate†
Received
21st December 2015
, Accepted 27th January 2016
First published on 5th February 2016
Abstract
The preparation and characterization of 7-(4-([2,2′:6′,2′′-terpyridin]-4′-yl)phenyl)-5,10,15,20-tetraphenylporphyrinatozinc(II), 3, are reported, and the structure of 3 has been confirmed by a single crystal structure determination. Reaction of RuCl3·3H2O with diethyl (4-([2,2′:6′,2′′-terpyridin]-4′-yl)phenyl)phosphonate, 4, followed by 3 in reducing conditions gives [Ru(3)(4)][PF6]2. In solution, 3 and [Ru(3)(4)][PF6]2 undergo two, reversible porphyrin-centred oxidation processes at lower potential than the Ru2+/Ru3+ process in [Ru(3)(4)][PF6]2. In the solution absorption spectra, the Soret and Q bands in 3 are little perturbed upon complex formation; the MLCT band in [Ru(3)(4)][PF6]2 has λmax = 492 nm. Spectroelectrochemical data for 3 and [Ru(3)(4)][PF6]2 are presented. [Ru(3)(4)]2+ binds to nanoparticulate TiO2 and the solid-state absorption spectrum confirms enhanced light absorption with respect to the standard dye-sensitized solar cell (DSC) dye N719. However, the photoconversion efficiencies of DSCs sensitized with [Ru(3)(4)]2+ are disappointingly low. Transient absorption spectroscopic studies on this series of compounds indicate that triplet–triplet energy transfer processes are likely to be responsible for this poor performance.
Introduction
Ever since the first development of Grätzel n-type dye-sensitized solar cells (DSCs) employing sintered semiconductor nanoparticles functionalized with ruthenium(II)-based dyes,1–3 efforts have been concentrated on the improvement of the photoconversion efficiency. At present, state-of-the-art photoconversion efficiencies approach ∼13% in DSCs using ruthenium(II)-based, metal-free organic and zinc(II) porphyrin-based dyes.4–16 Nature's reliance on porphyrins in photosystem II has lead to significant interest in bioinspired devices utilizing porphyrin or metalloporphyrin-based sensitizers in DSCs,4,17–19 and a power conversion efficiency of 13% has been reported by Grätzel and coworkers for a porphyrin dye incorporating a donor–π–bridge–acceptor domain combined with a cobalt(II)/(III)redox shuttle.14
While both ruthenium- and porphyrin-based sensitizers are popular choices in DSCs, to the best of our knowledge, few sensitizers have combined polypyridylruthenium(II) and porphyrin domains in a single molecular species.20–22 Our approach to ruthenium(II) dyes containing light-harvesting porphyrin domains is predicated upon heteroleptic {RuII(tpy)2} domains in which the two tpy ligands bear anchoring and porphyrin substituents, respectively. The majority of oligopyridine–porphyrin conjugates are characterized by the attachment of the metal-binding domains directly or with a spacer to the phenyl-substituents of 5,10,15,20-tetraphenyl-21H,23H-porphyrins.23
We have now developed new approaches for the functionalization of porphyrins in which oligopyridines and their metal complexes are attached directly to a pyrrole ring of the porphyrin core. The selective monobromination of H2TPP in the 7-position developed by Zhang and coworkers,24 provided an attractive opening for our synthetic investigations into mono-functionalization of a porphyrin core with a 2,2′:6′,2′′-terpyridine (tpy) domain.
Experimental
General
Microwave reactions were carried out in a Biotage Initiator 8 reactor. 1H, 13C and 31P NMR spectra were recorded at room temperature using a Bruker Avance III-500 NMR spectrometer. 1H and 13C NMR chemical shifts were referenced to residual solvent peaks with respect to δ(TMS) = 0 ppm and 31P NMR chemical shifts with respect to δ(85% aqueous H3PO4) = 0 ppm. Solution absorption and emission spectra were measured using an Agilent 8453 spectrophotometer and a Shimadzu RF-5301PC spectrofluorometer, respectively. Spectroelectrochemical and solid-state absorption spectroscopic measurements used a Varian-Cary 5000 spectrophotometer. Electrospray (ESI) mass spectra and high resolution ESI-MS were measured on Bruker Esquire 3000plus and Bruker maXis 4G instruments, respectively. Nanosecond transient absorption spectra were measured on an LP-920KS spectrometer from Edinburgh Instruments using a frequency-doubled Quantel Brilliant B laser as a pump source. Transient absorption measurements with picosecond time resolution were performed with the TRASS instrument from Hamamatsu, equipped with a C7701-01 streak camera. Excitation occurred with a picosecond mode-locked Nd:YVO4/YAG laser (PL2251B-20-SH/TH/FH) with PRETRIG option from Ekspla.
Electrochemical measurements were carried out using a CH Instruments 900B potentiostat with [nBu4N][PF6] (0.1 M) as supporting electrolyte and at a scan rate of 0.1 V s−1. The working electrode was glassy carbon, pseudo-reference electrode silver wire and counter-electrode platinum wire; potentials were referenced with respect to the Fc/Fc+ couple. Spectroelectrochemical measurements were performed using a CH2Cl2 solution of 3 (1 mM) and an MeCN solution of [Ru(3)(4)][PF6]2 (0.6 mM) at room temperature with [nBu4N][PF6] (≈0.1 M) as the supporting electrolyte. The solution was added to an optically transparent thin-layer electrochemical (OTTLE) cell with two Pt minigrid electrodes (working and auxiliary), a silver wire pseudoreference electrode, and a path length of ≈0.2 mm. The potential was controlled using a VersaSTAT 3 potentiostat from Princeton Applied Research.
RuCl3·3H2O was purchased from Oxkem, and 5,10,15,20-tetraphenyl-21H,23H-porphyrin (H2TPP), NBS and [Pd(PPh3)4] from Sigma-Aldrich and used as received. 7-Bromo-5,10,15,20-tetraphenyl-21H,23H-porphyrin,24 1,25 2 (ref. 26) and 4 (ref. 27) were prepared as previously reported and spectroscopic data matched those in the literature.
Compound 3
Compound 1 (100 mg, 0.13 mmol, 1 eq.), K2CO3 (72 mg, 0.52 mmol, 4 eq.) and 2 (53 mg, 0.15 mmol, 1.2 eq.) were combined in a 20 mL microwave vial and dissolved in a mixture of toluene (9.5 mL) and deionized water (0.6 mL). N2 was bubbled through the solution for 30 min and then [Pd(PPh3)4] (15 mg, 0.013 mmol, 0.1 eq.) was added and the vial sealed. The reaction mixture was heated for 4 h at 120 °C in a microwave reactor, after which time the solvent was removed under reduced pressure. Column chromatography (SiO2, CHCl3) was performed on the crude material. The fraction containing [Zn(TPP)] eluted first and compound 2 was collected as the second (dark red-purple) fraction (Rf = 0.1). 3 was isolated as a dark purple solid (89 mg, 0.090 mmol, 69%). 1H NMR (500 MHz, CDCl3) δ/ppm 8.95–8.99 (m, 2H, HF3+F4/F3′+F4′), 8.96 (s, 1H, HH3), 8.95 (s, 2H, HI3+I4), 8.89 (d, J = 4.5 Hz, 2H, HF3+F4/F3′+F4′), 8.84 (d, J = 4.5 Hz, 2H, HF3/F4/F3′/F4′), 8.79 (s, 2H, HB3), 8.76 (ddd, J = 4.7, 1.8, 0.9 Hz, 2H, HA6), 8.71 (dt, J = 8.0, 1.1 Hz, 2H, HA3), 8.28–8.23 (m, 6H, HE2+G2), 7.93–7.88 (m, 4H, HA4+D2), 7.79–7.71 (m, 11H, HE3+E4+G3+G4+C2), 7.50 (m, 2H, HC3), 7.37 (m, 2H, HA5), 7.23–7.29 (m, 3H, HD3+D4). 13C NMR (126 MHz, CDCl3) δ/ppm 156.6 (CA2/B2), 156.1 (CA2/B2), 151.4 (CQ), 150.7 (CQ), 150.49 (CQ), 150.47 (CQ), 150.43 (CQ), 150.42 (CQ), 150.4 (CB4), 149.3 (CA6), 148.0 (CQ), 146.85 (CQ), 146.8 (CQ), 143.0 (CQ), 142.91 (CQ), 142.89 (CQ), 141.5 (CQ), 140.8 (CQ), 137.0 (CD2/A4), 135.8 (CD2/A4), 135.4 (CH3/Q), 135.35 (CH3/Q), 134.6 (CE2/G2), 134.55 (CE2/G2), 132.9 (CF3/F4/F3′/F4′/I3/I4), 132.3 (CF3/F4/F3′/F4′/I3/I4), 132.25 (CF3/F4/F3′/F4′/I3/I4), 132.2 (CF3/F4/F3′/F4′/I3/I4), 132.1 (CF3/F4/F3′/F4′/I3/I4), 131.6 (CF3/F4/F3′/F4′/I3/I4), 131.0 (CC3), 127.7 (CE3/E4/G3/G4), 127.65 (CE3/E4/G3/G4), 127.3 (CD4), 126.8 (CE3/E4/G3/G4), 126.2 (CD3+C2), 123.9 (CA5), 122.6, 121.7 (CQ), 121.6 (CA3), 121.2 (CQ), 120.8 (CQ), 118.8 (CB3) (CQ = quaternary C, not unambiguously assigned). ESI MS m/z (positive mode) 984.7 [M + H]+ (calc. 984.3). UV-Vis (EtOH, 9.7 × 10−7 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1) 285 (56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 315 (41000), 427 (520000), 560 (26000), 599 (9500). Found C 75.89, H 4.41, N 9.49; C65H41N7Zn·3H2O: C 75.10, H 4.56, N 9.43.
000), 315 (41000), 427 (520000), 560 (26000), 599 (9500). Found C 75.89, H 4.41, N 9.49; C65H41N7Zn·3H2O: C 75.10, H 4.56, N 9.43.
[Ru(4)Cl3]
Compound 4 (27 mg, 0.061 mmol, 1 eq.) and RuCl3·3H2O (16 mg, 0.061 mmol, 1 eq.) were suspended in EtOH (10 mL) and heated at reflux for 3.5 h. A brown precipitate formed, which was centrifuged, then washed with EtOH and Et2O. Brown [Ru(4)Cl3] (28 mg, 0.043 mmol, 70%) was used for the next step without further characterization.
[Ru(3)(4)][PF6]2
[Ru(4)Cl3] (28 mg, 0.043 mmol, 1 eq.), 3 (42.4 mg, 0.043 mmol, 1 eq.) and N-ethylmorpholine (2 drops) were suspended in dry EtOH (2 mL) in a microwave reactor vial. The mixture was heated in a microwave reactor at 140 °C for 20 min and then the dark red solution was poured into saturated aqueous NH4PF6 (50 mL). The precipitate that formed was centrifuged down, collected and washed with water and Et2O (3 × 5 mL). The residue was dissolved in MeCN and purified by column chromatography (SiO2, MeCN/saturated aq. KNO3/water 7:1:0.5 by volume). The first orange fraction (Rf = 0.67) was collected, then concentrated to 3 mL and poured in saturated aqueous NH4PF6 (25 mL) to give a precipitate which was washed with water and EtOH (3 × 5 mL). [Ru(3)(4)][PF6]2 was isolated as a purple powder (20 mg, 0.011 mmol, 26%). 1H NMR (500 MHz, CD3CN) δ/ppm 9.06 (s, 2H, HB3′′), 9.01 (s, 2H, HB3), 8.89 (m, 2H, HF3+F4/F3′+F4′), 8.86 (s, 1H, HH3), 8.85 (s, 2H, HI3+I4), 8.82 (d, J = 4.5 Hz, 2H, HF3/F4/F3′/F4′), 8.79 (d, J = 4.5 Hz, 2H, HF3/F4/F3′/F4′), 8.75 (m, 2H, HA3′′), 8.69 (m, 2H, HA3), 8.33 (m, 2H, HC2′′), 8.30 (m, 2H, HG2), 8.24 (m, 4H, HE2), 8.09 (m, 2H, HC3′′), 7.98–8.05 (m, 6H, HA4′′+A4+D2), 7.95 (d, J = 8.5 Hz, 2H, HC2), 7.77–7.86 (m, 9H, HE3+E4+G3+G4), 7.70 (d, J = 8.5 Hz, 2H, HC3), 7.53 (m, 2H, HA6′′), 7.47 (m, 2H, HA6), 7.43–7.46 (m, 1H, HD4), 7.38 (m, 2H, HD3), 7.27 (m, 2H, HA5′′), 7.22 (m, 2H, HA5), 4.14 (m, 4H, HEt), 1.35 (t, J = 7 Hz, 6H, HEt). 13C NMR (126 MHz, CD3CN) δ/ppm 159.3 (CA2′′/B2′′), 159.0 (CA2/B2), 156.8 (CA2′′/B2′′), 156.3 (CA2/B2), 153.4 (CA6+A6′′), 151.9 (CQ), 151.0 (CQ), 148.5 (CQ), 147.9 (CB4), 147.1 (CB4′′), 147.0 (CQ), 144.0 (CQ), 143.8 (CQ), 143.2 (CQ), 143.0 (CQ), 141.5 (CQ), 139.0 (CA4+A4′′), 136.9 (CD2), 135.7 (CH3), 135.3 (CE2), 135.2 (CG2), 134.4 (CQ), 133.4 (CC3′′), 133.1 (CF3/F4/F3′/F4′), 132.7 (CI3+I4+two of F3/F4/F3′/F4′), 132.2 (HF3/F4/F3′/F4′), 132.1 (CC3), 131.8 (CC4′′, JPC = 189 Hz), 129.0 (CC2′′), 128.5 (HA5+A5′′), 127.8 (CE3+E4+G3+G4+D4), 127.4 (CC2), 127.0 (CD3), 125.5 (CA3+A3′′), 123.0 (CB3′′), 122.2 (CB3), 63.2 (CEt), 16.6 (CEt), (CQ = quaternary C, not all resolved). ESI MS m/z (positive mode) 765.6 [M]2+ (calc. 765.7), negative mode 144.8 [PF6]− (calc. 145.0). UV-Vis (MeCN, 1 × 10−6 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1) 284 (79000), 312 (87000), 425 (370000), 492 (56000), 559 (28000), 599 (10000). Found: C 57.38, H 4.07, N 8.01; C90H65F12N10O3P3RuZn·2H2O requires C 58.18, H 3.74, N 7.54.
Crystallography
Data were collected on a Bruker Kappa Apex2 diffractometer with data reduction, solution and refinement using APEX28 and CRYSTALS.29 The program Mercury v. 3.730,31 was used for structural analysis.
Compound 3·Me2CO
C68H47N7OZn, M = 1043.55, red block, monoclinic, space group P21/c, a = 13.1741(13), b = 20.850(3), c = 19.578(2) Å, β = 102.558(6)°, U = 5249.2(10) Å3, Z = 4, Dc = 1.320 Mg m−3, μ(Cu-Kα) = 1.063 mm−1, T = 123 K. Total 66771 reflections, 9749 unique, Rint = 0.078. Refinement of 7565 reflections (694 parameters) with I > 2σ(I) converged at final R1 = 0.0894 (R1 all data = 0.2457), wR2 = 0.1059 (wR2 all data = 0.2583), gof = 1.0000. CCDC 1442417.
DSC fabrication
Solaronix Test Cell Titania Electrodes were heated to 450 °C for 30 min and then cooled to 80 °C, when they were dipped into the dye-baths. N719 (Solaronix) reference electrodes and electrodes with adsorbed [Ru(3)(4)]2+ were made by dipping in an EtOH solution of N719 (0.1 mM) or MeCN solution of [Ru(3)(4)][PF6]2 (0.1 mM), respectively, for 3 days. The electrodes were taken out of the dye-baths, washed with EtOH or MeCN, respectively, and dried using a heatgun (60 °C). Commercial counter electrodes (Solaronix Test Cell Platinum Electrodes) were washed with EtOH, and then heated on a hot plate at 450 °C for 30 min to remove volatile organic impurities.
The working and counter-electrodes were joined using thermoplast hot-melt sealing foil (Solaronix Test Cell Gaskets, 60 μm) by heating while pressing together. The electrolyte (LiI (0.1 M), I2 (0.05 M), 1-methylbenzimidazole (0.5 M), 1-butyl-3-methylimidazolinium iodide (0.6 M) in 3-methoxypropionitrile) was inserted between the electrodes by vacuum backfilling through a hole in the counter electrode; this was sealed (Solaronix Test Cell Sealings) and capped (Solaronix Test Cell Caps). All DSCs were fully masked for measurements.32,33
Electrodes for solid-state absorption spectroscopy
Dye-functionalized electrodes were assembled as above but using Solaronix Test Cell Titania Electrodes Transparent.
DSC measurements
Masks for the DSCs were made from a black-coloured copper sheet with an aperture of average area 0.06012 cm2 (1% standard deviation) placed over the active area of the DSC. The area of the mask hole was less than the surface area of TiO2 (0.36 cm2). Black tape was used to complete the masking of the cell. Performance measurements were made by irradiating the DSC from behind with a LOT Quantum Design LS0811 instrument (100 mW cm−2 = 1 sun), and the simulated light power was calibrated with a silicon reference cell.
Results and discussion
Synthesis and characterization of compound 3
The synthetic route to the porphyrin-functionalized 2,2′:6′,2′′-terpyridine 3 is summarized in Scheme 1. The selective bromination of H2TPP in the 7-position was carried out using NBS as described by Zhang and coworkers,24 and an excess of zinc(II) acetate25 was added to yield zinc(II) complex 1. When the metallation of H2TPP with zinc(II) was carried out prior to reaction with NBS, selective halogenation was no longer observed and a mixture of brominated derivatives was obtained. The reaction of 1 with boronic acid 226 under Suzuki–Miyaura cross-coupling conditions led to 3 in 69% yield after workup. Metallation of the porphyrin core before the coupling reaction is essential. Although 7-bromo-5,10,15,20-tetraphenyl-21H,23H-porphyrin undergoes coupling with 2 to give the zinc-free analogue of 3, subsequent reaction with Zn(OAc)2·4H2O leads to competition between the porphyrin and tpy metal-binding domains for coordination to zinc(II). Thus, the sequence of steps presented in Scheme 1 is the optimal route to 3.
 |
| | Scheme 1 Synthetic route to 3. Conditions: (i) NBS, CHCl3 reflux, 4.5 h; (ii) 2 equivalents Zn(OAc)2 in MeOH; room temperature, 17 h, CHCl3; (iii) K2CO3, 2 in toluene/H2O, [Pd(PPh3)4], 4 h, 120 °C under microwave conditions. | |
The highest mass peak envelope in the electrospray mass spectrum of 3 came at m/z 984.7 and exhibited a characteristic isotope pattern for zinc. The 1H NMR spectrum of 3 is shown in Fig. 1a and was assigned using COSY and NOESY methods. The spectrum is consistent with the desymmetrization of the [Zn(TPP)] domain. This most noticeably affects the ortho-protons (HD2, HE2 and HG2) of the phenyl rings and the remaining protons in phenyl ring D (Fig. 1a). The shift to lower frequency of the signals for HD3 and HD4 compared to the meta- and para-protons in rings E and G is attributed to the proximity of HD3 and HD4 to the arene ring C. The 13C NMR spectrum of 3 was assigned using HMQC and HMBC methods (Fig. S1 and S2†).
 |
| | Fig. 1 Aromatic region of the 500 MHz NMR spectra of (a) 3 (in CDCl3) and (b) [Ru(3)(4)][PF6]2 (in CD3CN). * = residual CHCl3. Chemical shifts in δ/ppm. See Schemes 1 and 2 for atom labels. | |
Single crystals of 3·Me2CO were grown by slow evaporation of solvent from an acetone solution of 3. The acetone adduct of 3 (Fig. 2) crystallizes in the monoclinic space group P21/c. The {Zn(TPP)} unit is structurally as expected with the atom Zn1 lying only 0.14 Å out of the mean plane of the porphyrin N4-donor set; Zn–N bond distances and N–Zn–N bond angles are given in the caption to Fig. 2. The acetone molecule is axially coordinated and the Zn–O bond distance of 2.345(4) Å is within the range of observed for axial ketones in porphyrinato zinc(II) complexes.34 The twist angles between the planes of the phenyl rings with C3, C14, C25 and C36 and the porphyrin core are in the range 59.8 and 67.4°; for the phenyl ring containing C45, the corresponding angle is 61.9°, and the pyridine ring with N6 is then twisted through 32.5° with respect to the plane of the phenyl spacer. The similar twist angles for the arene rings containing C36 and C45 permit the rings to engage in a π-stacking interaction although the 16.2° angle between their planes is not ideal; the centroid⋯centroid distance is 3.51 Å. The tpy unit is virtually planar (angles between planes of adjacent pyridine rings are 2.7 and 6.2°). The planarity is associated with a face-to-face π-interaction between centrosymmetric pairs of tpy units (Fig. 3a). The centrosymmetric pairing of the {Zn(TPP)} units (Fig. 3b) is typical and has been extensively discussed in the literature.35
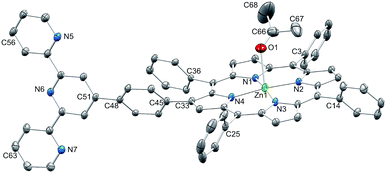 |
| | Fig. 2 Structure of 3·Me2CO with H atoms omitted for clarity; thermal ellipsoids plotted at 30% probability level. Selected bond metrics: Zn1–N1 = 2.034(3), Zn1–N2 = 2.062(3), Zn1–N3 = 2.042(3), Zn1–N4 = 2.062(3), Zn1–O1 = 2.345(4), C66–O1 = 1.215(8) Å; N1–Zn1–N2 = 89.69(14), N2–Zn1–N3 = 89.27(13), N1–Zn1–N4 = 89.52(13), N3–Zn1–N4 = 90.51(13), N1–Zn1–O1 = 82.10(16), N2–Zn1–O1 = 88.43(16), N3–Zn1–O1 = 106.45(16), N4–Zn1–O1 = 98.11(16)°. | |
 |
| | Fig. 3 (a) Stacking of centrosymmetric pairs of tpy domains in 3·Me2CO; separation of central pyridine rings = 3.30 Å and centroid⋯centroid distance = 3.71 Å. (b) Stacking of centrosymmetric pairs of {Zn(TPP)} units; Zn(1)⋯Zn(1)i = 6.903(1) Å (symmetry code i = 2 − x, 1 − y, 2 − z). | |
Synthesis and characterization of [Ru(3)(4)]PF6]2
Heteroleptic [Ru(tpy-I)(tpy-II)]2+ complexes are most conveniently made by treating RuCl3·3H2O sequentially with the two ligands, the second step in the presence of N-ethylmorpholine for the reduction of ruthenium(III) to ruthenium(II). In principle, two approaches could be used to prepare [Ru(3)(4)][PF6]2: (i) reaction of RuCl3·3H2O with 3, followed by treatment with 4 in the presence of N-ethylmorpholine, or (ii) reaction of RuCl3·3H2O with 4, followed by treatment with 3 in the presence of N-ethylmorpholine. Both methods were tried but the former resulted in cleavage of the porphyrin and tpy domains in 3. Thus, [Ru(3)(4)][PF6]2 was prepared as shown in Scheme 2 by treatment of RuCl3·3H2O with 4 to give the insoluble species [Ru(4)Cl3] which was further reacted with 3 in the presence of N-ethylmorpholine to give (after anion exchange and workup) [Ru(3)(4)][PF6]2 as a purple powder in 26% yield.
 |
| | Scheme 2 Synthesis of [Ru(3)(4)]2+, isolated as the [PF6]− salt. Conditions: RuCl3·3H2O, EtOH, reflux, 4.5 h; (ii) 2, EtOH, N-ethylmorpholine, 140 °C, 20 min under microwave conditions; (iii) NH4PF6. Atom labels for NMR spectra of [Ru(3)(4)][PF6]2 are shown. | |
The ESI mass spectrum of [Ru(3)(4)][PF6]2 showed a peak envelope at m/z 765.6 with peaks at half-mass intervals consistent with assignment to [Ru(3)(4)]2+. In addition to signals for aromatic protons, the 1H NMR spectrum of a CD3CN solution of [Ru(3)(4)][PF6]2 exhibited multiplets at δ 4.14 and 1.35 ppm, arising from the ethyl groups of the phosphonate ester group in ligand 4. The relative integrals of these resonances compared to the aromatic region confirmed that no ester hydrolysis occurred during the complex formation; partial hydrolysis of PO(OEt)2-functionalized tpy ligands has been observed during the formation of some ruthenium(II) complexes.36–38 The aromatic region of the [Ru(3)(4)][PF6]2 is shown in Fig. 1b and its signature is consistent with the presence of two different tpy domains. COSY, NOESY, HMQC and HMBC methods were used to assign the signals in the 1H and 13C NMR spectra, although not all quaternary signals could be unambiguously ascribed (see Experimental section). A starting point for distinguishing between the two tpy ligands was assignment of the ipso-C atom of the arene ring attached to the phosphonate group; the resonance for CC4′′ (Scheme 2) was a doublet (JPC = 189 Hz) at δ 131.8 ppm. A comparison of Fig. 1a with Fig. 1b shows that formation of the {Ru(tpy)2}2+ domain leads to the characteristic shift of the HA6 signal to lower frequency (δ 8.76 ppm in 3 to δ 7.47 ppm in [Ru(3)(4)][PF6]2); the signal for HA6′′ (δ 7.53 ppm) appears close to that for HA6, consistent with these protons lying over the ring current of the adjacent tpy ligand in the octahedral {Ru(tpy)2}2+ unit.
Cyclic voltammetry
The redox behaviour of [Zn(TPP)] has previously been described,39 and the potentials for the reversible one-electron reduction and oxidation processes provide reference data for the interpretation of the electrochemical behaviour of the new conjugate species. Data are summarized in Table 1 along with the electrochemical band-gaps, ΔE1/2. The electrochemistry of 3 and [Ru(3)(4)][PF6]2 was studied using cyclic voltammetry. Like [Zn(TPP)], compound 3 exhibits two oxidative processes (Table 1 and Fig. 4a), reversible and reproducible over three scans, which are assigned to [Zn(TPP)]/[Zn(TPP)]+ and [Zn(TPP)]+/[Zn(TPP)]2+ couples. Reduction processes were poorly defined in the CV (Fig. S3†) but could be distinguished using differential pulse voltammetry; Ered1/2 values are given in Table 1. By comparison with data for [Zn(TPP)], the first reduction at −1.76 V is presumably centred on the {Zn(TPP)} domain in 3 and the processes at more negative potential are phenyltpy-centred. On going to [Ru(3)(4)][PF6]2, the potential of the [Zn(TPP)]-centred oxidative couples are little affected (Table 1). An additional oxidation process at +0.88 V (Table 1 and Fig. 4b) is assigned to the Ru2+/Ru3+ couple, and compares well with +0.895 V reported for [Ru(Phtpy)2][PF6]2.40 [Ru(3)(4)][PF6]2 undergoes three irreversible reduction processes.
Table 1 Redox potentials for 3 in CH2Cl2 and [Ru(3)(4)][PF6]2 in MeCN solution compared to [Zn(TPP)] in CH2Cl2 measured using cyclic voltammetry. Potentials are referenced to Fc/Fc+ with 0.1 M [nBu4][PF6] as supporting electrolyte and a scan rate of 0.1 V s−1 (ir = irreversible, qr = quasi-reversible)a
| Compound |
Eox1/2/V |
Ered1/2/V |
ΔE1/2/Vb |
Reference |
| Values in parenthesis = Epc − Epa in mV. ΔE1/2 = Eox1/2 − Ered1/2 |
| [Zn(TPP)] |
+0.42 (82) |
−1.79 (79) |
2.21 |
39 |
| +0.71 (81) |
| 3 |
+0.34 (57) |
−1.76ir |
2.10 |
This work |
| +0.66 (84) |
−1.97ir |
| |
−2.09ir |
| |
−2.27ir |
| [Ru(3)(4)][PF6]2 |
+0.39 (56) |
−1.56ir |
1.95 |
This work |
| +0.67 (56) |
−1.87ir |
| +0.88 (65) |
−2.21ir |
 |
| | Fig. 4 Oxidative processes in the cyclic voltammograms of (a) 3 and (b) [Ru(3)(4)][PF6]2. For conditions, see Table 1. | |
Absorption spectra and spectroelectrochemistry
Fig. 5 compares the solution absorption spectra of 1, 3 and [Ru(3)(4)][PF6]2. The spectrum of compound 1 shows the typical features of a metallated porphyrin,39,41 the intense Soret band at 425 nm (ε = 660000 dm3 mol−1 cm−1) arising from the S2 ← S0 transition, and weaker Q bands resulting from the vibrational structure ((0, 0) and (0, 1)) from the S1 ← S0 transition (558 and 597 nm, ε = 22000 and 7600 dm3 mol−1 cm−1, respectively). On going to compound 3, an additional π-conjugated system in the β-pyrrolyl position is introduced. The Soret band moves slightly and decreases in intensity (427 nm, ε = 520000 dm3 mol−1 cm−1), while the Q bands change little (Fig. 5, 560 and 599 nm, ε = 26000, 9500 dm3 mol−1 cm−1, respectively). The high-energy bands (around 285 nm, green trace in Fig. 4) are attributed to π* ← π transitions localized on the phenyltpy domain.
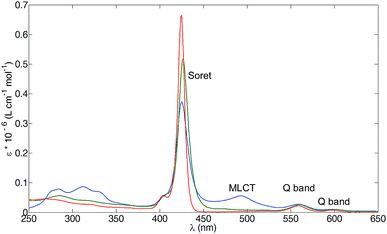 |
| | Fig. 5 Absorption spectra of 1 (red line, EtOH, λmax = 425 nm), 3 (green line, EtOH, λmax = 427 nm) and [Ru(3)(4)][PF6]2 (blue line, MeCN, λmax = 425 nm). Concentration = 1 × 10−6 mol dm−3. | |
Upon formation of the ruthenium(II) complex [Ru(3)(4)][PF6]2, the high-energy bands (below 350 nm) approximately doubled in intensity with respect to the absorptions in 3 (blue trace in Fig. 5), consistent with the presence of two tpy domains. The Soret band again decreases in intensity (ε = 370000 dm3 mol−1 cm−1) but is little shifted from the free ligand 3 (425 versus 427 nm), providing evidence for electronic communication between the porphyrin and tpy domains. Fig. 5 shows that there is also little difference in the Q bands comparing 3 with [Ru(3)(4)][PF6]2, (in the complex, 599 and 559 nm, ε = 28000, 10000 dm3 mol−1 cm−1, respectively). Confirmation of the presence of the {Ru(tpy)2}2+ chromophore comes from the appearance of the broad band at 492 nm arising from the 1MLCT absorption of this chromophore.42
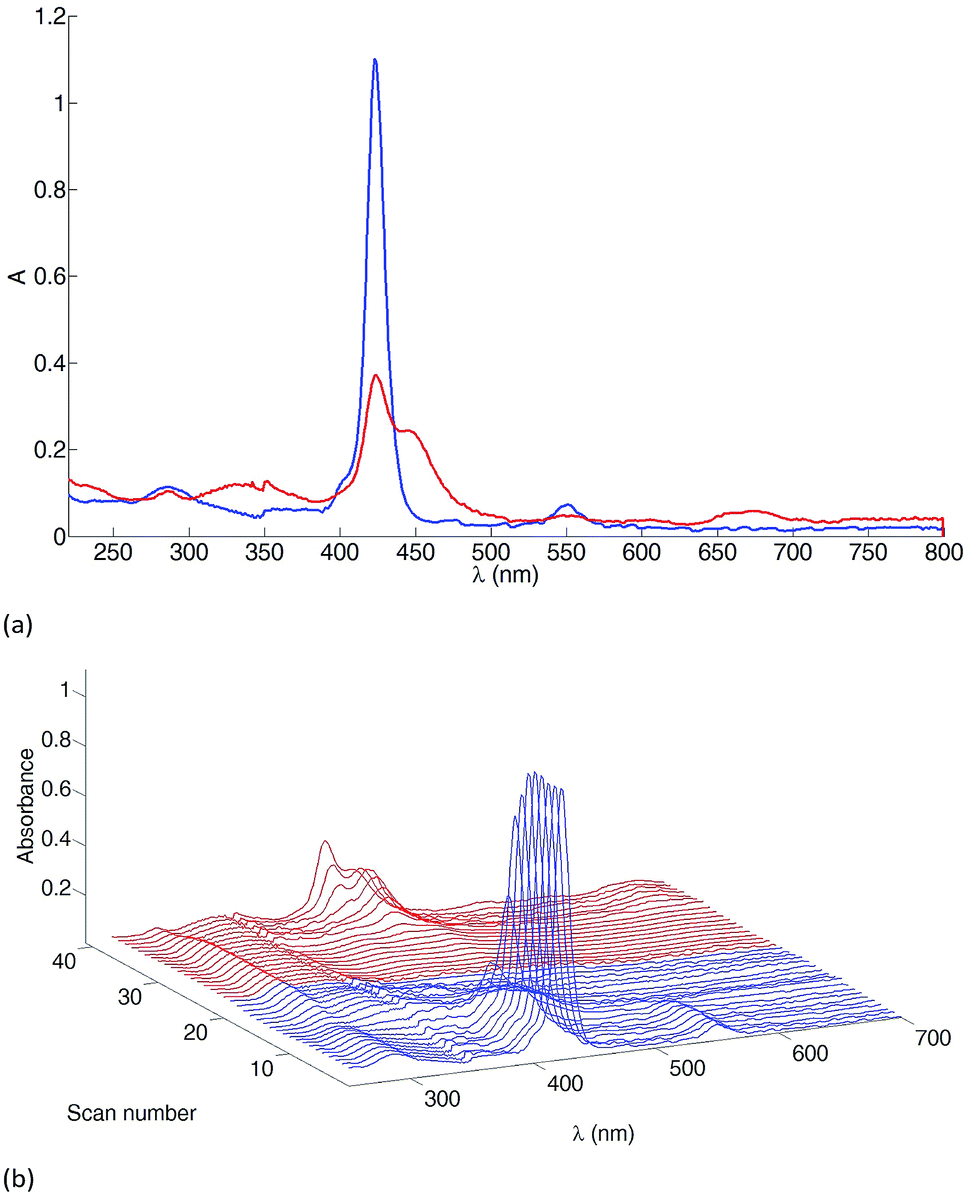
We commence the spectroelectrochemical discussion by presenting the results obtained from a study of a CH2Cl2 solution of compound 3. Fig. 6a shows a superimposition of the absorption spectra of 3 recorded before and after an oxidative cycle. The complete set of scans for the oxidative cycle are shown in Fig. 6b. The irreversible changes are consistent with the literature data for [Zn(TPP)].43 Initial oxidation leads to a π-radical cation, the Soret band of which is about half as intense as the original band, and the Q bands are replaced with an absorption with λmax ≈ 650 nm. The second oxidation leads to a dication which is unstable on the experimental timescale. The results of the spectroelectrochemical reductive cycle for 3 are depicted in Fig. S4† and are again consistent with the literature data for [Zn(TPP)].
 |
| | Fig. 6 Spectroelectrochemical data for the oxidative cycle of 3 (≈1 mM in CH2Cl2, [nBu4N][PF6] supporting electrolyte). (a) Absorption spectra before (blue line) and after (red line) the oxidative cycle. (b) A spectrum was recorded every 0.1 V, starting from 0 V (first blue line at the front) to +1.8 V (last blue line) and back from +1.8 V (first red line) to 0 V (last red line). The potential is referenced with respect to the Fc/Fc+ redox couple with the same cell under the same experimental conditions. | |
The oxidative and reductive cycles of the spectroelectrochemical measurements carried out on an MeCN solution of [Ru(3)(4)][PF6]2 are shown in Fig. 7 and 8. At the end of the oxidative cycle, the regeneration of the absorptions associated with the {Ru(tpy)2}2+ domain (the MLCT band at 492 nm, and the bands at 284 and 310 arising from the phenyltpy π* ← π transitions) confirms the reversibility of these processes. In contrast, the oxidation of the [Zn(TPP)] moiety within the complex is irreversible, the processes mimicking those of compound 3 with the exception that the band at 650 nm is now transient. This may be due to over-oxidation of the porphyrin core. During the reductive cycle (Fig. 8), the absorptions arising from both the porphyrin and {Ru(tpy)2}2+ domains are irreversibly transformed. The irreversible changes to the Soret and Q bands are consistent with those observed for 3 and [Zn(TPP)]43 while irreversible reduction processes centred on the phenyltpy units are responsible for the loss of the bands associated with the π* ← π and MLCT transitions. Fig. 9b provides evidence for a transient band between 800 and 900 nm, which can be assigned to the [3]˙− radical anion.44
 |
| | Fig. 7 Spectroelectrochemical data for the oxidative cycle of [Ru(3)(4)][PF6]2 (≈1 mM in MeCN, [nBu4N][PF6] supporting electrolyte). (a) Absorption spectra before (blue line) and after (red line) the oxidative cycle. (b) A spectrum was recorded every 0.1 V, starting from 0 V (first blue line at the front) to +1.5 V (last blue line) and back from +1.5 V (first red line) to 0 V (last red line). See caption to Fig. 6 for referencing to Fc/Fc+. | |
 |
| | Fig. 8 Spectroelectrochemical data for the reductive cycle of [Ru(3)(4)][PF6]2 (≈1 mM in MeCN, [nBu4N][PF6] supporting electrolyte). (a) Absorption spectra before (blue line) and after (red line) the reductive cycle. (b) A spectrum was recorded every 0.1 V, starting from 0 V (first blue line at the front) to −1.8 V (last blue line) and back from −1.8 V (first red line) to 0 V (last red line). See caption to Fig. 6 for referencing to Fc/Fc+. | |
 |
| | Fig. 9 Solid-state absorption spectra of transparent TiO2 electrodes with dyes N719 (red) and [Ru(3)(4)][PF6]2 (blue). | |
[Ru(3)(4)][PF6]2 as a dye in DSCs
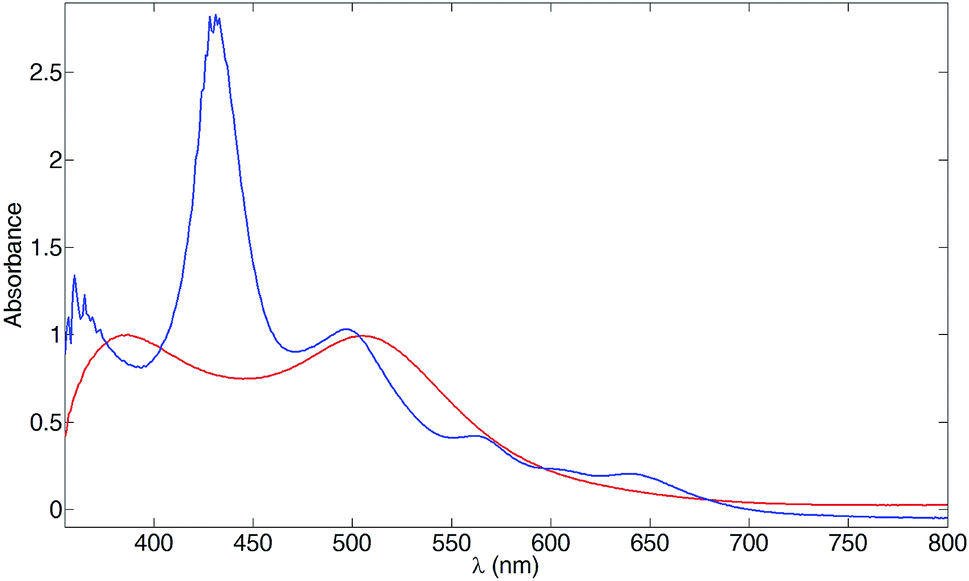
Although phosphonic acid anchors45 bind more strongly than phosphonate esters,46 it is has been demonstrated that TiO2 surfaces can be functionalized using phosphonate esters47,48 with immobilization of the anchor taking place by hydrolysis of POR groups by surface-OH groups.49 We therefore investigated the use of [Ru(3)(4)]2+ as a dye in DSCs. First, we confirmed that the dye bound to a TiO2 surface. TiO2 electrodes (without a scattering layer) were soaked in an MeCN solution of [Ru(3)(4)][PF6]2 for 3 days, and were then washed and dried. The electrode retained a red colour similar to that of reference electrodes with adsorbed N719. Compared to N719, the additional spectral response that the Soret band imparts to [Ru(3)(4)]2+ is clear from the solid-state absorption spectra Fig. 9. Adsorbed [Ru(3)(4)]2+ exhibits λmax at 432, 500, 564 and 641 nm. The Soret band at 432 nm is red-shifted with respect to solution (425 nm) and absorptions at 500, 564 and 641 nm compare with bands in the solution spectrum (Fig. 5) at 492 nm (MLCT) and 560 and 600 nm (Q bands).
Photoanodes for n-type DSCs were made by immersion of FTO/TiO2 electrodes in an MeCN solution of [Ru(3)(4)][PF6]2 for 3 days, and reference electrodes were made similarly using an EtOH solution of N719. DSCs were fabricated using an I−/I3− electrolyte (see Experimental section) and were fully masked.32,33 The reproducibility of performance parameters (Table 2) was confirmed using duplicate DSCs for each dye. Despite the enhanced light absorption of [Ru(3)(4)]2+ with respect to N719, the conversion efficiency (η) is poor; the main contributing factor is the extremely low short-circuit current density (JSC) electron injection. The open-circuit voltage (VOC) is about half that of N719. In order to understand the poor performance of [Ru(3)(4)]2+ in DSCs, we have carried out a detailed investigation of the energy-transfer processes that follow excitation.
Table 2 Performance parameters of duplicate DSCs with [Ru(3)(4)]2+ and N719. Measurements were made on the day of DSC fabrication
| Dye |
JSC/mA cm−2 |
VOC/mV |
ff/% |
η/% |
| N719 (cell 1) |
12.29 |
657 |
67 |
5.37 |
| N719 (cell 2) |
11.21 |
655 |
65 |
4.74 |
| [Ru(3)(4)]2+ (cell 1) |
0.10 |
333 |
52 |
0.02 |
| [Ru(3)(4)]2+ (cell 2) |
0.11 |
334 |
53 |
0.02 |
Emission properties
The solution emission behaviour of 3 and [Ru(3)(4)][PF6]2 were investigated and compared to those of 1. As discussed earlier, in the absorption spectrum of the latter, bands arising from S2 ← S0 and S1 ← S0 transitions are observed. Normally, for an organic molecule, population of the S2 excited state is followed by fast internal conversion to S1,50 and the emission spectrum can be related to the radiative decay of the lowest excited state of same multiplicity. [Zn(TPP)] is emissive from both the S2 and S1 states, although the fluorescence originating from the S2 state has a much lower quantum yield and only a picosecond lifetime.51 Excitation of 1 at 400 nm (into the Soret shoulder) results in the emission spectrum shown in Fig. 10 with fluorescence from both the S2 (431 and 453 nm) and S1 (607 and 659 nm) excited states. The assignments were confirmed from the excitation spectra. Note that λmax of the Soret band (425 nm) is too close to the λmaxem of 431 and 453 nm from the S2 fluorescence to observe these emissions using λexc = 425 nm. Excitation into the Q bands of 1 leads to the S1 emissions at 607 and 659 nm.
 |
| | Fig. 10 Solution emission spectrum of 1 (EtOH, 1 × 10−6 M, room temperature). λexc = 400 nm. | |
The emission spectrum of compound 3 does not display an S2 fluorescence. Excitation into either the Soret or Q bands leads to emission at 613 and 660 nm. Interestingly it is possible to detect porphyrin fluorescence even upon exciting into the tpy bands (λexc = 285 and 320 nm). Since the tpy absorption is well separated from the porphyrin absorption bands, this is a clear indication of intramolecular energy transfer. The energetics of the system are favourable for an energy transfer from the (π–π*) tpy excited states (upper lying levels) to the S2 state (lower level), followed by internal conversion to S1 and radiative decay. Excitation spectra monitored at 560 and 600 nm confirm the presence of a broad peak centred at 285 nm, in agreement with the involvement of a tpy absorption in the population of the S1 state. The ruthenium(II) complex [Ru(3)(4)][PF6]2 exhibits an emission behaviour similar to that of 3. Excitation into the tpy absorption bands (λexc = 284 and 310 nm) results in porphyrin fluorescence (λmaxem = 613 and 661 nm). Moreover Fig. 11 shows that if the excitation is in the MLCT band (λexc = 492 nm), emission is again observed from the S1 state of the porphyrin. It was not possible to detect S2 fluorescence by exciting into the shoulder of the Soret band.
 |
| | Fig. 11 Solution emission spectrum of [Ru(3)(4)][PF6]2 (MeCN, 1 × 10−6 M, room temperature). λexc = 492 nm. | |
Transient absorption spectra of [Ru(3)(4)][PF6]2
In order to further probe the emission behaviour of [Ru(3)(4)][PF6]2, the transient absorption spectrum of the complex was recorded at room temperature. Upon excitation in the MLCT band (λexc = 532 nm), the transient absorption spectrum obtained resembles the characteristic triplet–triplet spectrum of [Zn(TPP)], first predicted by Gouterman52 and later reported by Holten and coworkers.53 Gouterman predicted two possible transitions from the porphyrin T1 state: an intense allowed transition that would result in a doubly excited configuration and consist of two absorption peaks to lower energy of the Soret band, and a weak, forbidden transition in the near infrared (IR) leading to a highly excited singlet configuration. In the transient absorption spectrum of [Ru(3)(4)][PF6]2 (Fig. 12), the loss of the ground state porphyrin is clearly indicated by bleaching of the Soret band (425 nm) and of the Q(0, 1) and Q(0, 0) bands (560 and 600 nm). The broad bands at 470 and 500 nm are associated with absorption of the porphyrin T1 state and creation of the doubly excited configuration. A broad absorption is present at lower energies, extending from 600 nm to the NIR, in agreement with the literature spectrum.53
 |
| | Fig. 12 (a) Transient absorption spectrum of [Ru(3)(4)][PF6]2 (MeCN, ≈2 × 10−6 M, room temperature). λexc = 532 nm. Acquisition time 200 ns, 5 acquisitions without time delay. (b) Temporal evolution of the optical density between 440 and 463 nm after excitation at 532 nm with laser pulses of 30 ps duration. | |
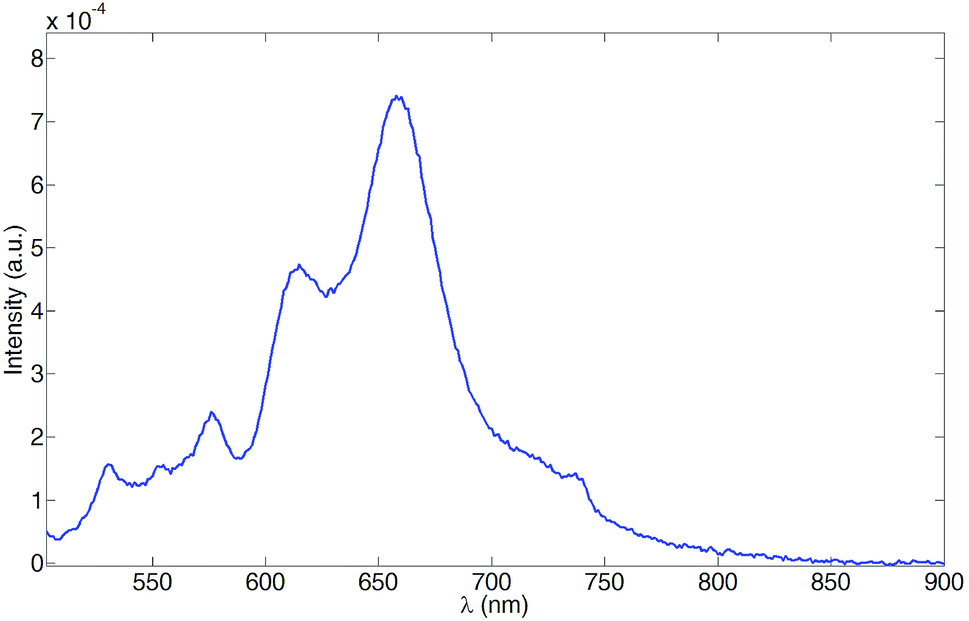
The near-IR (NIR) transient absorption spectra of [Ru(3)(4)][PF6]2 are shown in Fig. 13. The band at 820 nm is associated with the forbidden, higher energy singlet which appears at 832 nm for [Zn(TPP)]53 in CH2Cl2. For [Ru(3)(4)][PF6]2, the NIR absorption was detected only in deaerated solution. The recovery of the ground state was monitored at 385, 425 and 470 nm for the aerated solution and at 385, 425, 470 and 820 nm for the deaerated one. As expected for a triplet state, the lifetimes ranged from hundreds of nanoseconds for the aerated solution to tens of microseconds for the deaerated one, due to the exclusion of a non-radiative deactivation pathway through reaction with triplet O2. The decay curves are consistent with a mono-exponential decay in all cases; the lifetimes for ground state recovery are: τ385 = 441 ± 44 ns and 59 ± 6 μs, τ425 = 418 ± 42 ns and 52 ± 5 μs, τ470 = 435 ± 44 ns and 48 ± 5 μs, τ820 = 49 ± 5 μs. Since all the lifetimes are consistent within experimental error, it is reasonable to assume that all observed transitions originate from a single chemical species which we propose to be the porphyrin T1 state. Furthermore, we conclude that upon MLCT excitation a triplet-to-triplet energy transfer occurs from the 3MLCT level to T1, the latter being the lowest accessible level for [Ru(3)(4)][PF6]2.
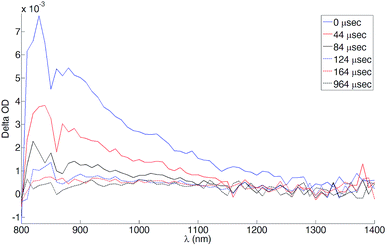 |
| | Fig. 13 NIR transient absorption spectra of [Ru(3)(4)][PF6]2 (MeCN, ≈2 × 10−6 M, room temperature). λexc = 532 nm. The spectra were reconstructed from decay curves recorded every 10 nm. | |
The literature contains a number of molecular triads and dyads related to [Ru(3)(4)][PF6]2 (Scheme 3).54 When the triad (Scheme 3a) reported by Benniston et al.55 is excited in the MLCT band, the 3MLCT emission is quenched in favour of a triplet-to-triplet energy transfer to the porphyrin T1 state. The lifetime of T1 was determined to be 65 ± 5 μs with a triplet-to-triplet energy transfer rate constant of 8 × 1010 s−1. A Dexter type mechanism of energy transfer56 was proposed, and it is significant that excitation into the Q band resulted in a decreased S1 fluorescence. Benniston et al.55 rationalize this in terms of singlet (S1) to triplet (3MLCT) energy transfer (k = 4 × 108 s−1), involving an endergonic Dexter type mechanism (a process that is spin-forbidden). In the osmium-containing dyad in Scheme 3b, excitation in the Soret band leads to direct transfer to the 3MLCT state57,58 followed by triplet-to-triplet energy transfer to T1. A high rate constant for porphyrin fluorescence quenching again accounts for complete energy transfer. In oligopyridine complexes, the excited MLCT state is localized on one of the ligands,59 and Benniston et al. argue that the energy flows from S1 to the tpy domain directly connected to the porphyrin. In addition, if the second tpy in the complex lacks an extended π-system in the 4-position of the central pyridine ring, the electronic energy can be considered to reside on the porphyrin-bearing tpy because the intra-ligand energy transfer would not be as fast as that of energy transfer to the porphyrin triplet state. The overall effect is an intersystem crossing involving the porphyrin unit, involving the {Ru(tpy)}2+ domain. For the triad reported by Flamigni et al. (Scheme 3c),60 the energy transfer rate constant for the triplet-to-triplet transfer is >5 × 1010 s−1, consistent with a fast and quantitative quenching of the ruthenium-containing manifold. Once again the fluorescence originating from S1 in quenched in favour of population of the 3MLCT.
 |
| | Scheme 3 Triads and dyads related to [Ru(3)(4)][PF6]2 and reported by (a) and (b) Benniston et al.,55,57 and (c) Flamigni et al.60 | |
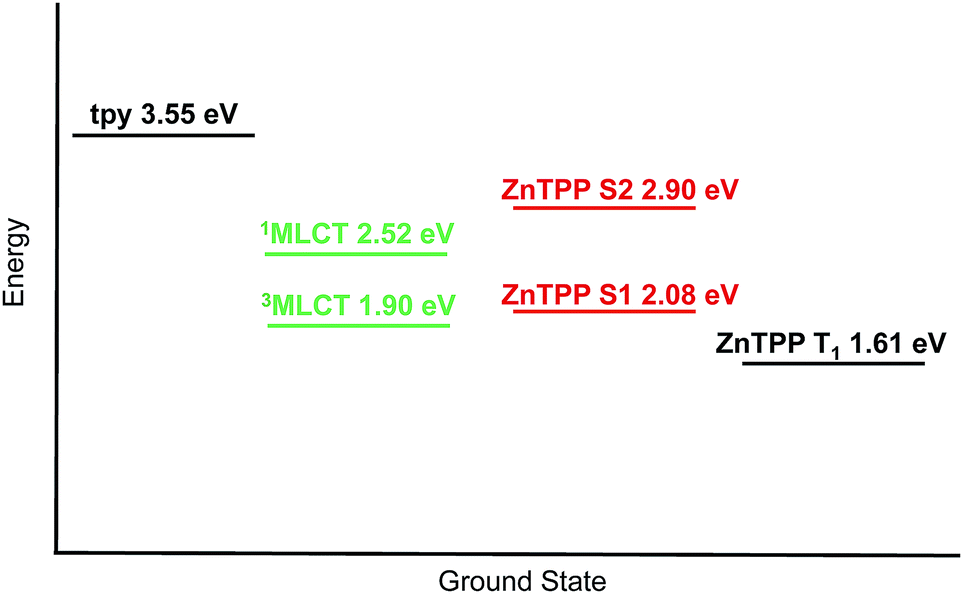
Fig. 14 shows the relative energies of the tpy, MLCT and {Zn(TPP)} singlet and triplet states of [Ru(3)(4)]2+. The energy of the tpy domain was obtained by plotting the normalized absorption and emission spectra (λexc = 284 and 310 nm) of [Ru(3)(4)][PF6]2, expressed in wavenumbers and searching for a crossing point which corresponds to the (0, 0) transition. The values obtained were 345 and 353 nm, corresponding to 3.59 and 3.51 eV. An average value of 3.55 eV has been adopted for the tpy level. The {Zn(TPP)} S2 level was obtained with the aforementioned procedure using spectra of 1. A value of 427 nm (2.90 eV) was obtained. The {Zn(TPP)} S1 level was calculated from spectra of [Ru(3)(4)][PF6]2, and searching for the crossing point between the Q(0, 0) bands. This gave a value of 596 nm (2.08 eV). The 1MLCT energy level was derived from the UV-Vis maximum of [Ru(3)(4)][PF6]2, and the 3MLCT level from the emission maximum of the analogous [Ru(pytpy)2][PF6]2 (pytpy = 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine) in MeCN (655 nm,61 1.89 eV) since the 3MLCT state of [Ru(3)(4)][PF6]2 is not emissive at room temperature in solution. Finally, the {Zn(TPP)} T1 energy (770 nm62 corresponding to 1.61 eV) was obtained from the literature data. The relative energies of the states allow us to propose the energy transfer process shown in Scheme 4. Upon 1MLCT excitation, fast intersystem crossing to 3MLCT occurs. Triplet-to-triplet energy transfer takes place with a rate constant >2 × 1010 s−1 (Fig. 14b), leading to the {Zn(TPP)} T1 state. By deactivation of this state the ground state is recovered.
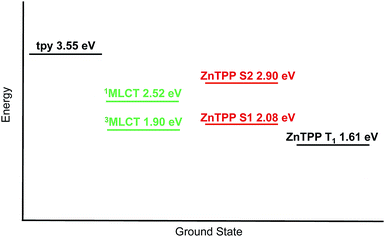 |
| | Fig. 14 Energetic level diagram for [Ru(3)(4)]2+. | |
 |
| | Scheme 4 Energy-transfer scheme upon 1MLCT excitation at 532 nm in [Ru(3)(4)][PF6]2. For the middle species in the scheme, the negative charge could be on either tpy domain. ISC = intersystem crossing. | |
Conclusions
We have prepared and characterized the porphyrin-functionalized tpy ligand 3, and its single crystal structure has been determined. The ruthenium(II) complex [Ru(3)(4)][PF6]2 contains the peripheral light-harvesting domain in 3 coupled with a phosphonate ester functionality in 4 that allows the complex to be bound to nanoparticulate TiO2, the n-type semiconductor applied on the photoanode in DSCs. In solution, 3 and [Ru(3)(4)][PF6]2 undergo two, reversible porphyrin-centred oxidation processes at lower potential than the Ru2+/Ru3+ couple in [Ru(3)(4)][PF6]2. In the solution absorption spectra, the Soret and Q bands in 3 are little affected upon coordination to ruthenium(II) and detailed spectroelectrochemical studies of 3 and [Ru(3)(4)][PF6]2 have been described. FTO/TiO2 electrodes have been functionalized with [Ru(3)(4)]2+ and solid-state absorption spectra demonstrate enhanced light absorption with respect to the standard DSC dye N719. However, the photoconversion efficiencies of dye-sensitized solar cells (DSCs) sensitized with [Ru(3)(4)]2+ are disappointingly low. Transient absorption spectroscopic studies indicate that triplet–triplet energy transfer processes are the most likely reason for the poor DSC photoconversion efficiencies.
Acknowledgements
We acknowledge the Swiss National Science Foundation as part of the NCCR Molecular Systems Engineering, and the University of Basel for financial support. The SNF R'Equip program is acknowledged for grant number 206021_157687/1. Nathalie Marinakis and Sarah Keller are thanked for providing ligand 4 and recording NMR spectra, respectively.
Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed; M. Grätzel, Inorg. Chem., 2005, 44, 6841 CrossRef PubMed; M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145 CrossRef and references therein.
- Dye Sensitized Solar Cells, ed. K. Kalyanasundaram, CRC Press, Boca Raton, 2010 Search PubMed.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Hagfeldt and M. Grätzel, Acc. Chem. Res., 2000, 33, 269 CrossRef CAS PubMed.
- Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Mishra, M. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C.-h. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103 CrossRef CAS PubMed.
- H. Ozawa, Y. Okuyama and H. Arakawa, ChemPhysChem, 2014, 15, 1201 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, T. Kyomen and M. Hanaya, Chem. Commun., 2015, 51, 6315 RSC.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS PubMed.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601 CrossRef CAS.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330 CrossRef CAS PubMed.
- C. Stangel, K. Ladomenou, G. Charalambidis, M. K. Panda, T. Lazarides and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2013, 1275 CrossRef CAS.
- I. Jung, H. Choi, J. K. Lee, K. H. Song, S. O. Kang and J. Ko, Inorg. Chim. Acta, 2007, 360, 3518 CrossRef CAS.
- A. L. A. Parussulo, B. A. Iglesias, H. E. Toma and K. Araki, Chem. Commun., 2012, 48, 6939 RSC.
- M. Schwalbe, P. Wrzolek, G. Lal and B. Braun, Eur. J. Inorg. Chem., 2014, 4209 CrossRef CAS; M. Schwalbe, R. Metzinger and T. S. Teets, Chem.–Eur. J., 2012, 18, 15449 CrossRef PubMed; C. Monnereau, J. Gomez, E. Blart, F. Odobel, S. Wallin, A. Fallberg and L. Hammarström, Inorg. Chem., 2005, 44, 4806 CrossRef PubMed.
- G.-Y. Gao, J. V. Ruppel, D. B. Allen, Y. Chen and X. P. Zhang, J. Org. Chem., 2007, 8, 9060 CrossRef PubMed.
- S. A. Vail, D. I. Schuster, D. M. Guldi, M. Isosomppi, N. Tkachenko, H. Lemmetyinen, A. Palkar, L. Echegoyen, X. Chen and J. Z. H. Zhang, J. Phys. Chem. B, 2006, 110, 14155 CrossRef CAS PubMed.
- H. S. Mehr, N. C. Romano, R. Altamimi, J. M. Modarelli and D. A. Modarelli, Dalton Trans., 2015, 44, 3176 RSC.
- V. Spampinato, N. Tuccitto, S. Quici, V. Calabrese, G. Marletta, A. Torrisi and A. Licciardello, Langmuir, 2010, 26, 8400 CrossRef CAS PubMed.
- APEX2, version 2 User Manual, Bruker Analytical X-ray Systems, Inc., M86-E01078, Madison, WI, 2006 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- H. J. Snaith, Energy Environ. Sci., 2012, 5, 6513 Search PubMed.
- H. J. Snaith, Nat. Photonics, 2012, 6, 337 CrossRef CAS.
- See for example: A. K. D. Dime, C. H. Devillers, H. Cattey, B. Habermeyer and D. Lucas, Dalton Trans., 2012, 41, 929 RSC; R. K. Kumar, A. Goldborth and I. Goldberg, Z. Kristallogr., 1997, 212, 383 Search PubMed; M. P. Bryn, C. J. Curtis, Yu. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480 CrossRef CAS; L. C. Gilday, N. G. White and P. D. Beer, Dalton Trans., 2012, 41, 7092 RSC.
- See for example: W. R. Scheidt and Y. J. Lee, Struct. Bonding, 1987, 64, 1 CrossRef CAS.
- S. M. Zakeeruddin, M. K. Nazeeruddin, P. Pechy, F. P. Rotzinger, R. Humphry-Baker, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1997, 36, 5937 CrossRef CAS PubMed.
- D. K. Zhong, S. Zhao, D. E. Polyansky and E. Fujita, J. Catal., 2013, 307, 140 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. Šmídková and J. A. Zampese, Can. J. Chem., 2014, 92, 724 CrossRef CAS.
- N. Armaroli, F. Diederich, L. Echegoyen, T. Habicher, L. Flamigni, G. Marconi and J.-F. Nierengarten, New J. Chem., 1999, 77 RSC.
- E. C. Constable, A. M. W. Cargill Thompson, D. A. Tocher and M. A. M. Daniels, New J. Chem., 1992, 16, 855 CAS.
- A. Harriman, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 1978 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- A. Klein, in Spectroelectrochemistry, ed. W. Kaim and A. Klein, Royal Society of Chemistry, Cambridge, 2008, ch. 4, p. 91 Search PubMed.
- J. Pawlik, L. Gherghel, S. Karabunarliev and M. Baumgarten, Chem. Phys., 1997, 221, 121 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427 CAS.
- F. J. Malzner, S. Y. Brauchli, E. Schönhofer, E. C. Constable and C. E. Housecroft, Polyhedron, 2014, 82, 116 CrossRef CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367 CrossRef CAS.
- G. Guerrero, P. H. Mutin, E. Framery and A. Vioux, New J. Chem., 2008, 32, 1519 RSC.
- A. Vioux, J. Le Bideau, P. H. Mutin and D. Leclercq, Top. Curr. Chem., 2004, 232, 145 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14 RSC.
- A. Lukaszewicz, J. Karolczak, D. Kowalska, A. Maciejewski, M. Ziolek and R. P. Steer, Chem. Phys., 2007, 331, 359 CrossRef CAS.
- M. Gouterman, J. Chem. Phys., 1960, 33, 1523 CrossRef CAS.
- J. Rodriguez, C. Kirmaier and D. Holten, J. Am. Chem. Soc., 1989, 111, 6500 CrossRef CAS.
- P. K. Poddutoori, P. Poddutoori and B. G. Maya, J. Porphyrins Phthalocyanines, 2006, 10, 1049 CrossRef CAS.
- A. C. Benniston, A. Harriman, C. Pariani and C. A. Sams, Phys. Chem. Chem. Phys., 2006, 8, 2051 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- A. C. Benniston, A. Harriman, C. Pariani and C. A. Sams, J. Phys. Chem. A, 2007, 111, 8918 CrossRef CAS PubMed.
- A. C. Benniston and A. Harriman, Coord. Chem. Rev., 2008, 252, 2528 CrossRef CAS.
- P. G. Bradley, N. Kress, B. A. Hornberger, R. F. Dallinger and W. H. Woodruff, J. Am. Chem. Soc., 1981, 103, 7441 CrossRef CAS.
- L. Flamigni, F. Barigelletti, N. Armaroli, B. Ventura, J.-P. Collin, J.-P. Sauvage and J. A. G. Williams, Inorg. Chem., 1999, 38, 661 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, A. Cargill Thompson, P. Passaniti, S. Silvi, M. Maestri and A. Credi, Inorg. Chim. Acta, 2007, 360, 1102 CrossRef CAS.
- V. A. Waiters, J. C. de Paula, B. Jackson, C. Nutaitis, K. Hall, J. Lind, K. Cardozo, K. Chandran, D. Raible and C. M. Phillips, J. Phys. Chem., 1995, 99, 1166 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2: HMQC and HMBC spectra of 3; Fig. S3 and S4: additional CV and spectroelectrochemical data for 3. CCDC 1442417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra27397h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a