
Access to indenofurans and indenopyridines via annulation of heterocyclic ketene aminals, o-phthalaldehyde and cyclic 1,3-diketones†
Feilong Sun,
Xusheng Shao* and
Zhong Li
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China. E-mail: shaoxusheng@ecust.edu.cn
First published on 1st February 2016
Abstract
An efficient and concise method was developed for access to indenofurans and indenopyridines through a one pot, three-component protocol from heterocyclic ketene aminals, o-phthalaldehyde and 1,3-diketones under catalyst-free conditions. The indenofurans and indenopyridines were formed via aldol condensation, a regioselective aza–ene reaction, imine–enamine tautomerization and intramolecular cyclization.
Introduction
Indenoheterocycle skeletons (Fig. 1) are widely distributed in a large number of natural products and synthetic molecules, showing a wide range of attractive biological properties.1 In particular, indenofurans and indenopyridines are of great interest due to their broad-ranging biological activity, such as anti-HIV activity,2 cytotoxicity,3 phosphodiesterase inhibition,4 anti-inflammatory,5 calcium modulating activities6 and adenosine A2a receptor antagonists.7 Because of their importance as privileged scaffolds in drug discovery, the construction of indenofurans8 and indenopyridines9 has been pursued intensively in recent years. However, most of the current synthetic methods are limited by their use of toxic catalysts and solvents, narrow substrate scope, harsh reaction conditions and operational complexity. Several benign protocols have been applied to construct indenopyridine derivatives recently,10 but developing novel synthetic methods are highly desirable. | ||
| Fig. 1 Bioactive indenoheterocycle derivatives. | ||
As a type of versatile building block for the synthesis of fused heterocycles, heterocyclic ketene aminals (HKAs) received much attention within the chemical community.11 HKAs have the highly polarized ethylene system with electron-donating amino group and electron-withdrawing substituent at two ends (Scheme 1). As a result, the α-carbon and the secondary amino groups usually react with bis-electrophiles to produce the fused heterocyclic compounds.12 2-(2-Chloroaroyl)methyleneimidazolidines (Scheme 1, I) as a novel HKA with four reactive sites have been well investigated by Li's group.13 It shows fascinating structural features of a polarized push–pull C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and chlorine atom as a leaving group (Scheme 1a). Shortly afterwards, Lin's group reported a new reactive site by incorporating the two nucleophilic sites N1 and C3 and the electrophile site C4 (CO) through a one-pot protocol via the cascade reaction to form polycyclic frame (Scheme 1b).14 Our recent interest focus on the reaction behaviors of HKAs II with nitro as electron-withdrawing group. Besides its two nucleophilic sites similar with that of I, nitro group can further participate in the cycloaddition reactions. In continuation of our efforts on HKAs chemistry,15 we herein discovered a novel reaction site of II that nitro-group as leaving group (Scheme 1c).
C double bond and chlorine atom as a leaving group (Scheme 1a). Shortly afterwards, Lin's group reported a new reactive site by incorporating the two nucleophilic sites N1 and C3 and the electrophile site C4 (CO) through a one-pot protocol via the cascade reaction to form polycyclic frame (Scheme 1b).14 Our recent interest focus on the reaction behaviors of HKAs II with nitro as electron-withdrawing group. Besides its two nucleophilic sites similar with that of I, nitro group can further participate in the cycloaddition reactions. In continuation of our efforts on HKAs chemistry,15 we herein discovered a novel reaction site of II that nitro-group as leaving group (Scheme 1c).
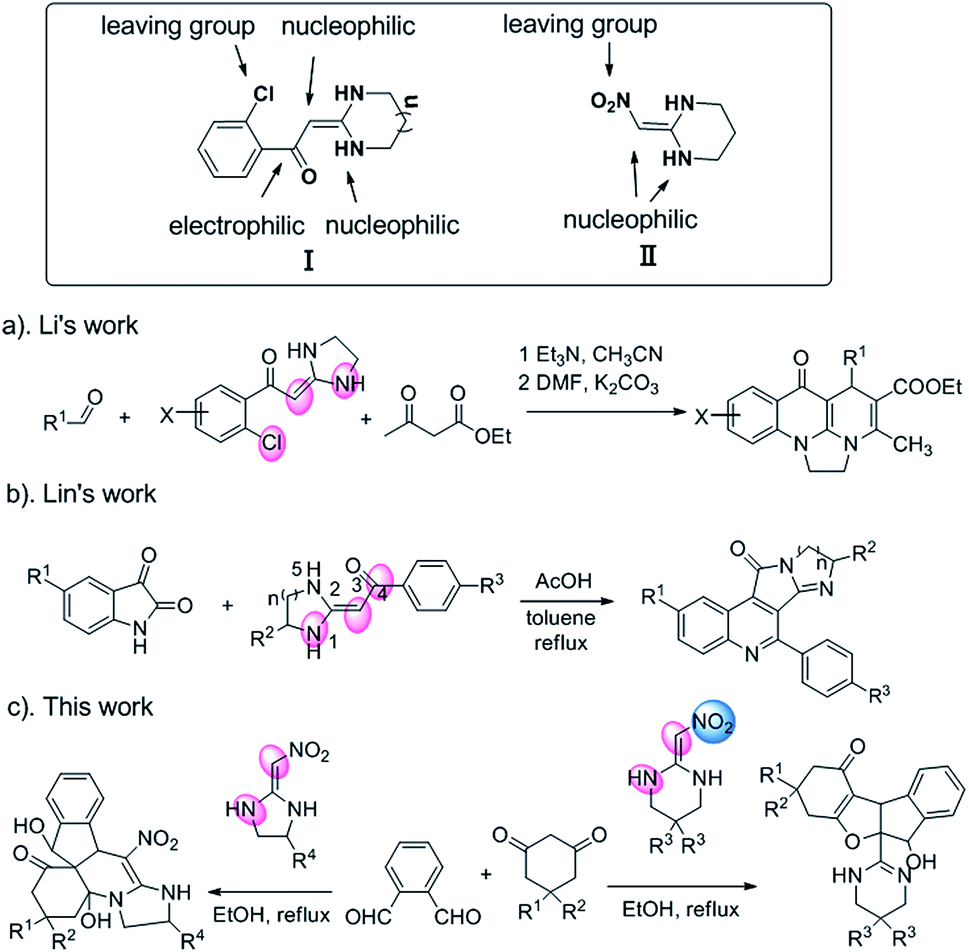 | ||
| Scheme 1 Reaction sites of HKAs. | ||
Results and discussion
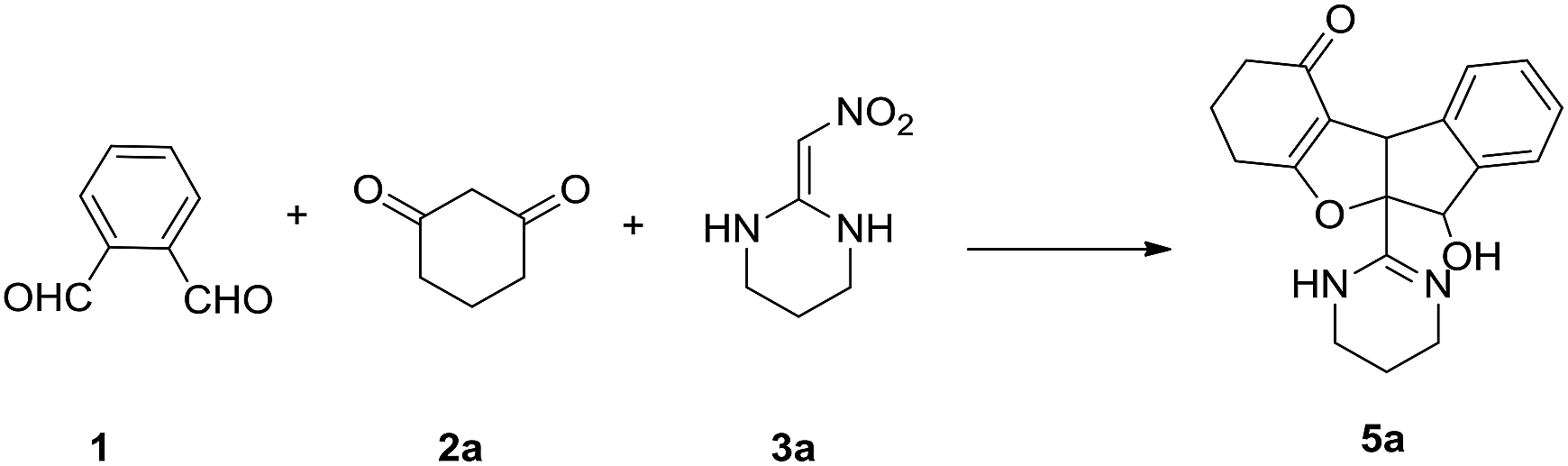
Ortho-phthalaldehyde (OPA) has proven to be an appealing building block for the reaction of their 1,4-biselectrophilic centers16 and widely used to detect the amino group.17 In view of the importance of HKAs and OPA, we turned our attention to develop a new strategy to construct novel heterocycles with the utilization of such two versatile synthonsWhen exploring the reaction behavior of HKA 3a, cyclohexane-1,3-dione 2a and OPA 1, we were delighted to find the reaction proceed smoothly affording 5a in 72% yield in the absence of catalyst under reflux in ethanol (Table 1, entry 1). Therefore, we selected this three-component domino reaction as the model reaction to explore the optimum reaction conditions. Initially, the molar ratio was screened. The experiments clearly demonstrated that the best molar ratio of 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 of 1:2a:3a (Table 1, entries 1–4). Then, the reaction was conducted using a 1.2:1:1 mixture of 1, 2a and 3a under various conditions. Subsequently, temperature evaluation showed that reflux was the best (Table 1, entries 2 and 5–7). In order to promote the reaction, different catalysts were examined. The addition of catalysts such as acetic acid (AcOH) and p-toluene sulfonic acid (p-TsOH), trimethylamine (Et3N), piperidine and L-proline reduced has detrimental effect on the reaction (Table 1, entries 8–12). Different solvents were evaluated suggesting ethanol was the optimal selection (Table 1, entries 13–17). From various entries displayed in Table 1, the green and efficient strategy was ethanol as the solvent in the absence of catalyst under reflux for 5 h (Table 1, entry 3).
:1:1 of 1:2a:3a (Table 1, entries 1–4). Then, the reaction was conducted using a 1.2:1:1 mixture of 1, 2a and 3a under various conditions. Subsequently, temperature evaluation showed that reflux was the best (Table 1, entries 2 and 5–7). In order to promote the reaction, different catalysts were examined. The addition of catalysts such as acetic acid (AcOH) and p-toluene sulfonic acid (p-TsOH), trimethylamine (Et3N), piperidine and L-proline reduced has detrimental effect on the reaction (Table 1, entries 8–12). Different solvents were evaluated suggesting ethanol was the optimal selection (Table 1, entries 13–17). From various entries displayed in Table 1, the green and efficient strategy was ethanol as the solvent in the absence of catalyst under reflux for 5 h (Table 1, entry 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Molar ratio (1a:2a:3) |
Catalyst | Temp (°C) | Time (h) | Yielda (%) |
| a UPLC yield based on HKA 3a.b Isolated yields based on HKA 3a. | ||||||
| 1 | EtOH | 1:1:1 |
— | Reflux | 5 | 72 |
| 2 | EtOH | 1:1:1.1 |
— | Reflux | 5 | 87 |
| 3 | EtOH | 1:1:1.2 |
— | Reflux | 5 | 89(77)b |
| 4 | EtOH | 1:1:1.3 |
— | Reflux | 5 | 76 |
| 5 | EtOH | 1:1:1.2 |
— | 60 | 6.5 | 82 |
| 6 | EtOH | 1:1:1.2 |
— | 40 | 9 | 80 |
| 7 | EtOH | 1:1:1.2 |
— | rt | 36 | Trace |
| 8 | EtOH | 1:1:1.2 |
AcOH | Reflux | 5 | 75 |
| 9 | EtOH | 1:1:1.2 |
p-TSA | Reflux | 5 | 38 |
| 10 | EtOH | 1:1:1.2 |
Et3N | Reflux | 6 | 59 |
| 11 | EtOH | 1:1:1.2 |
Piperidine | Reflux | 6 | 58 |
| 12 | EtOH | 1:1:1.2 |
L-Proline | Reflux | 6 | 60 |
| 13 | CH3CN | 1:1:1.2 |
— | Reflux | 5 | 77 |
| 14 | CH3OH | 1:1:1.2 |
— | Reflux | 5 | 82 |
| 15 | H2O | 1:1:1.2 |
— | Reflux | 5 | 58 |
| 16 | i-Propanol | 1:1:1.2 |
— | Reflux | 5 | 81 |
| 17 | CHCl3 | 1:1:1.2 |
— | Reflux | 5 | 82 |
With the optimal reaction condition in hand, we gained insight into the tolerance of this method. We investigated the generality of this transformation with respect to various cyclic 1,3-diketone compounds and six-membered HKAs (Scheme 2). Four representative cyclic 1,3-diketones were used to react with HKA 3a (Scheme 2, 5a, 5c, 5e and 5g). Cyclic 1,3-diketones bearing different substituents could smoothly give the target products as mixture of stereoisomers in good yields (70–85%). The products 5a and 5c showed good diastereocontrol with a ratio 22:1 and 18:1,‡ respectively. In contrast, 5e and 5g exhibited poor diastereoselectivity. In an endeavor to expand the scope of six-membered HKAs, HKA 3b was reacted with four 1,3-diketones as mentioned above. Compared to 3a, 3b rendered lower yields probably due to steric hindrance (Scheme 2, 5b, 5d, 5f and 5h), following with poor diastereoselectivity except 5b and 5d.
 | ||
| Scheme 2 Access of indenofuran derivatives 5. | ||
In order to further demonstrate the generality of this method, we expanded it to five-membered HKAs 4a-b. Unexpectedly, the indenopyridine derivate 6a was formed in excellent yields (84%) in ethanol under reflux for 1 h, albeit without the desired indenofuran analogues of 5 (Scheme 3, 6a). Different products (5 vs. 6) was obtained with different membered HKAs (3 vs. 4) and this phenomenon can be attributed to the diverse regioselective nucleophilicity of α-carbon which was resulted from the ring size of HKAs (Scheme 5). With the obtaining of this interesting structure, we then directly explored the substrate scope and the intriguing regioselective phenomenon under conditions as same as HKAs 3a-b. To our delight, HKAs 4a-b reacted smoothly with the aforementioned cyclic 1,3-diketones giving indenopyridine derivatives 6 in good yields (Scheme 3, 6a–h). HKA 4b bearing a methyl group at the five-membered cycle exhibited lower yield than the substrate 4a, which is similar with the phenomenon observed on the six-membered HKAs. Unfortunately, all the reactions of the five-membered HKAs provided products 6 with poor diastereoselectivity except 6a. The steric hindrance generated by the substituents in 1,3-cyclohexanedione accounted partly for this phenomenon. Moreover, the substituents at the five-membered HKAs have great influence on the diasteteroselectivity.18 Then we explored the 1,3-cyclopentanedione having smaller ring size and found that the reaction proceeded smoothly affording products 6i-j in good yields, albeit with poor diastereoselectivity (Scheme 3, 6i-j). The tension of cyclic 1,3-diketones has little effect on the reaction with five-membered HKAs. Based on such fascinating observation, we extended the reaction to 1,3-indanedione, affording the desired products 8a-b (Scheme 4, 8a-b). Subsequently, we were curious about the reaction behavior of six-membered HKAs could with 1,3-indanedione. Surprisingly, indenopyridine derivatives 8c-d was generated instead of indenofurans in good yields with poor diastereoselectivity (Scheme 4, 8c-d). Notably, neither indenofurans nor indenopyridines were obtained with respect to the reactions between six-membered HKAs and 1,3-cyclopentanedione.
 | ||
| Scheme 3 Synthesis of indenopyridine derivatives 6. | ||
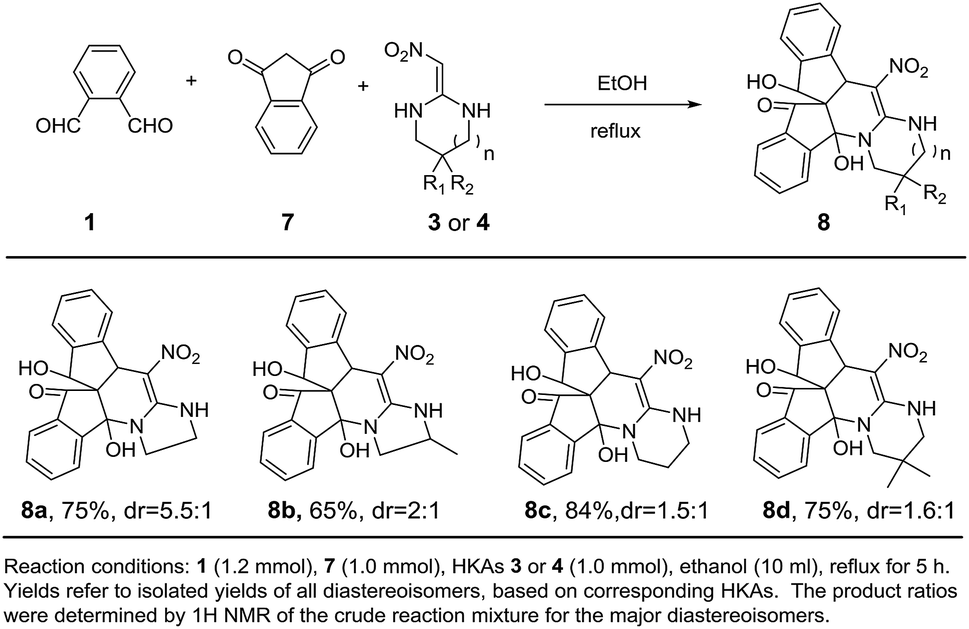 | ||
| Scheme 4 Synthesis of indenopyridine derivatives 8. | ||
To verify the structures of the indenofuran and indenopyridine derivatives, 5a and 6b were characterized by X-ray crystallography studies (ESI†). The structures of all products were well characterized by NMR and HRMS studies.
Upon watching the reaction, we occasionally found that white precipitate generated and disappeared during the preparation of product 5c at 40 °C in ethanol. Thus, the crude product was filtrated and analyzed by the LC-MS, affording two molecular weight 399.2 and 355.2 which were accordance with the corresponding intermediate C (or D) (Scheme 5) and 5c, respectively.
 | ||
| Scheme 5 Mechanism hypothesis for the construction of indenofurans and indenopyridines. | ||
Based on the above results and related literature,19–21 a tentative reaction mechanism is proposed as depicted in Scheme 2. Firstly, OPA undergoes aldol condensation with 5, 5-dimethylcyclohexane-1,3-dione 2b to afford intermediate A, followed by intramolecular dehydration to yield intermediate B. Then two different directions occur for the six- and five-membered HKAs. HKA 3a reacts with the aldehyde group of B and affords intermediate C via an aza–ene addition reaction and C undergoes a rapid imine–enamine tautomerization to give intermediate D. Then intramolecular aza–ene addition and cyclization occur, affording E. Finally, intermediate enolate nucleophilic attacking occurs and nitro-group leaves, giving the final product 5c. In contrast, five-membered HKA 4a undergoes nucleophilic addition to CC bond of intermediate B via an aza–ene addition reaction and cyclization, affording F. The intermediate G is then converted into final indenopyridines 6c via intramolecular cyclization.
Conclusions
In summary, we developed an efficient one-pot three components reaction to synthesize novel indenofuran and indenopyridine derivatives under catalyst-free condition. The reaction that α-carbon undergoes such regioselective reaction resulting from the ring sizes of HKAs was firstly reported. For five-membered HKAs' reaction: one C–O bond, two new rings and three C–C bonds were constructed. On the other hand, for six-membered HKAs: one C–N bond, two new rings and three C–C bonds were prepared. This strategy includes some advantages, such as high regioselectivity, the absence of catalysts and good diastereoselectivity for some components. The process presented a method for the construction of substituted indenofurans and indenopyridines with nitro and phenyl group, which could be transformed into new functionalities. Moreover, the investigation of the products' insecticidal activity is underway in our group and we hope this strategy may be of value for other researchers seeking novel biological molecules.Experimental
1H NMR and 13C NMR spectra were recorded on BrukerAM-400 (1H at 400 MHz, 13C at 100 MHz) spectrometer with DMSO-d6 as the solvent and TMS as the internal standard. Chemical shifts are reported in δ (parts per million) values. High resolution electron mass spectra (ESI-TOF) were performed on a Micromass LC-TOF spectrometer. Analytical thin-layer chromatography (TLC) was carried out on precoated plates (silica gel 60 F254), and spots were visualized with ultraviolet (UV) light. Chromatographic analysis was performed using an ACQUITY UPLC-H Class system (Waters Corp., USA), equipped with HSS T3 reversed phase column with 100 mm × 2.1 mm i.d. and 1.8 μm particle size, equipped with a quaternary solvent delivery system, a 48-vial autosampler (10 μL loop), and a photodiode array detector (PDA). The UPLC separations were carried out using gradient separation at a flow rate of 0.4 mL min−1. The mobile phase was a mixture of MilliQ ultrapure water with 0.01% trifluoroacetic acid (A) and acetonitrile (B). The following elution gradient totally lasted 15 min: initial mobile-phase composition, 90:10 (v/v) phase A:B; 0–8 min, linear change from 10 to 100% B; 8–10 min 100% B; 10–11 min, 90:10 (v/v) phase A:B. The column and injection chamber were maintained at 40 and 25 °C, respectively. The sample injection volume was 2 μL and the detector was set at 261 nm. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad singlet, coupling constant (Hz) and integration. In this paper, yields refer to isolated yields of all diastereoisomers based on HKAs and the ratios of major diastereoisomers were determined by 1H NMR of the crude reaction mixture.
Reagents
Heterocyclic ketene aminals (HKAs) were synthesized according to the reported procedures,15a all other solvents and reagents were purchased directly from commercial suppliers and used as received without further purification.General procedure for the synthesis of products 5
o-Phthalaldehydes 1 (1.2 mmol), cyclic 1,3-dicarbonyl compounds 2 (1.0 mmol), HKAs 3 (1.0 mmol) and ethanol (10 mL) were placed in a 25 mL round-bottom flask and the mixture was stirred under reflux for 5 h. After completion of the reaction (confirmed by TLC), the reaction mixture was filtered to afford the product or the solvent was evaporated and the product was obtained by crystallization from dichloromethane.General procedure for the synthesis of products 6 and 8
o-Phthalaldehydes 1 (1.2 mmol), cyclic 1,3-dicarbonyl compounds 2 or 7 (1.0 mmol), HKAs 3 (1.0 mmol) and ethanol (10 mL) were placed in a 25 mL round-bottom flask and the mixture was stirred under reflux for 1 h or 4 h. After completion of the reaction (confirmed by TLC), the reaction mixture was filtered to afford the crude product. One representative diastereoisomer was obtained by recrystallization from 95% EtOH.Acknowledgements
This work was financial supported by National Natural Science Foundation of China (21372079, 21472046), Shanghai Pujiang Program (14PJD012) and the Fundamental Research Funds for the Central Universities (222201414015), this work was also partly supported by Australia DC Foundation.Notes and references
- (a) D. S. Kim, N. I. Baek, S. R. Oh, K. Y. Jung, I. S. Lee and H. K. Lee, Phytochemistry, 1997, 46, 177–178 CrossRef CAS; (b) I. K. Bae, H. Y. Min, A. R. Han, E. K. Seo and S. K. Lee, Eur. J. Pharmacol., 2005, 513, 237–242 CrossRef CAS PubMed; (c) M. Nagai, S. Nagumo, S. M. Lee, I. Eguchi and K. I. Kawai, Chem. Pharm. Bull., 1986, 34, 1–6 CrossRef CAS; (d) R. L. Tolman and A. C. Chin, WO 20010193864, 2001; (e) W. Mar, H. T. Lee, K. H. Je, H. Y. Choi and E. K. Seo, Arch. Pharmacal Res., 2003, 26, 147–150 CrossRef CAS; (f) C. Nebot, M. Moutet, P. Huet, J. Z. Xu, J. C. Yadan and J. Chaudiere, Anal. Biochem., 1993, 214, 442–451 CrossRef CAS PubMed; (g) J. Xu and J. C. Yadan, Tetrahedron Lett., 1996, 37, 2421–2424 CrossRef CAS; (h) D. A. Nugiel, A.-M. Etzkorn, A. Vidwans, P. A. Benfield, M. Boisclair, C. R. Burton, S. Cox, P. M. Czerniak, D. Doleniak and S. P. Seitz, J. Med. Chem., 2001, 44, 1334–1336 CrossRef CAS PubMed; (i) R. Frédérick, W. Dumont, F. Ooms, L. Aschenbach, C. J. Van der Schyf, N. Castagnoli, J. Wouters and A. Krief, J. Med. Chem., 2006, 49, 3743–3747 CrossRef PubMed.
- W. L. Xiao, L. M. Yang, N. B. Gong, L. Wu, R. R. Wang, J. X. Pu, X. L. Li, S. X. Huang, Y. T. Zheng, R. T. Li, Y. Lu, Q. T. Zheng and H. D. Sun, Org. Lett., 2006, 8, 991–994 CrossRef CAS PubMed.
- R. Miri, K. Javidnia, B. Hemmateenejad, A. Azarpira and Z. Amirghofran, Bioorg. Med. Chem., 2004, 12, 2529–2536 CrossRef CAS PubMed.
- G. R. Heintzelman, K. M. Averill and J. H. Dodd, PCT Int. Appl., WO2002085894 A1 20021031, 2002.
- K. Cooper, M. J. Fray, P. E. Cross and K. Richardson, Eur. Pat. Appl., EP 299727 A1 19890118, 1989.
- C. Safak, R. Simsek, Y. Altas, S. Boydag and K. Erol, Boll. Chim. Farm., 1997, 136, 665–669 CAS.
- G. R. Heintzelman, K. M. Averill, J. H. Dodd, K. T. Demarest, Y. Tang and P. F. Jackson, Pat. Appl. Publ., US 2004082578 A1 20040429, 2004.
- (a) G. Bianchini, A. Cavarzan, A. Scarso and G. Strukul, Green Chem., 2009, 11, 1517–1520 RSC; (b) S. Ozaki, H. Matsushita and H. Ohmori, J. Chem. Soc., Chem. Commun., 1992, 1120–1122 RSC; (c) Y. Miller, L. Miao, A. S. Hosseini and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 12149–12156 CrossRef CAS PubMed.
- (a) D. Tadić, B. K. Cassels, A. Cavé, M. O. F. Goutlart and A. B. De Oliveira, Phytochemistry, 1987, 26, 1551–1552 CrossRef; (b) N. S. Prostakov, A. T. Soldatenkov, P. K. Radzhan, V. O. Fedorov, A. A. Fomichev and V. A. Rezakov, Chem. Heterocycl. Compd., 1982, 18, 390–394 CrossRef; (c) A. Padwa, T. M. Heidelbaugh and J. T. Kuethe, J. Org. Chem., 2000, 65, 2368–2378 CrossRef CAS PubMed; (d) T. Alves, A. B. de Oliveira and V. Snieckus, Tetrahedron Lett., 1988, 29, 2135–2136 CrossRef CAS; (e) K. Van Emelen, T. De Wit, J. Hoornaert and F. Compernolle, Tetrahedron, 2002, 58, 4225–4236 CrossRef CAS; (f) N. D. Smith, J. Hayashida and V. H. Rawal, Org. Lett., 2005, 7, 4309–4312 CrossRef CAS PubMed.
- (a) M. Manpadi, P. Y. Uglinskii, S. K. Rastogi, K. M. Cotter, Y.-S. Wong, L. A. Anderson, A. J. Ortega, S. Van slambrouck, W. F. A. Steelant, S. Rogelj, P. Tongwa, M. Y. Antipin, I. V. Magedov and A. Kornienko, Org. Biomol. Chem., 2007, 5, 3865–3872 RSC; (b) X. Feng, J. J. Wang, Z. Xun, Z. B. Huang and D. Q. Shi, J. Org. Chem., 2015, 80, 1025–1033 CrossRef CAS PubMed.
- K. M. Wang, S. J. Yan and J. Lin, Eur. J. Org. Chem., 2014, 1129–1145 CrossRef CAS.
- (a) J. Fan, Q. Y. Yang, G. J. He, X. G. Xie, H. Y. Zhu, Y. Jin and J. Lin, RSC Adv., 2014, 4, 28852–28855 RSC; (b) B. Zhou, Z. C. Liu, W. W. Qu, R. Yang, X. R. Lin, S. J. Yan and J. Lin, Green Chem., 2014, 16, 4359–4370 RSC.
- L.-R. Wen, C. Liu, M. Li and L. J. Wang, J. Org. Chem., 2010, 75, 7605–7614 CrossRef CAS PubMed.
- F. Yu, S. Yan, L. Hu, Y. Wang and J. Lin, Org. Lett., 2011, 13, 4782–4785 CrossRef CAS PubMed.
- (a) F. Sun, X. Shao and Z. Li, Synlett, 2015, 26, 2306–2312 CrossRef CAS; (b) N. Chen, M. Zou, X. Tian, F. Zhu, D. Jiang, J. Cheng, X. Shao and Z. Li, Eur. J. Org. Chem., 2014, 28, 6210–6218 CrossRef; (c) R. B. Xu, R. Xia, M. Luo, X. Y. Xu, J. G. Cheng, X. S. Shao and Z. Li, J. Agric. Food Chem., 2014, 62, 381–390 CrossRef CAS PubMed; (d) Z. J. Ye, L. N. Shi, X. S. Shao, X. Y. Xu, Z. P. Xu and Z. Li, J. Agric. Food Chem., 2013, 61, 312–319 CrossRef CAS PubMed; (e) S. Lu, X. Shao, Z. Li, Z. Xu, S. Zhao, Y. Wu and X. Xu, J. Agric. Food Chem., 2012, 60, 322–330 CrossRef CAS PubMed.
- (a) C. Navarro and A. G. Csákÿ, Org. Lett., 2008, 10, 217–219 CrossRef CAS PubMed; (b) B. Jiang, B. M. Feng, S. L. Wang, S. J. Tu and G. Li, Chem.–Eur. J., 2012, 18, 9823–9826 CrossRef CAS PubMed.
- P. Zuman, Anal. Lett., 2005, 38, 1213–1220 CrossRef CAS.
- F. Yu, R. Huang, H. Ni, J. Fan, S. Yan and J. Lin, Green Chem., 2013, 15, 453–462 RSC.
- X. T. Zhu, H. W. Xu, B. Jiang, J. Y. Liu and S. J. Tu, Tetrahedron Lett., 2013, 54, 6341–6344 CrossRef CAS.
- X. B. Chen, Z. C. Liu, X. R. Lin, R. Huang, S. J. Yan and J. Lin, ACS Sustainable Chem. Eng., 2014, 2, 2391–2398 CrossRef CAS.
- W. Ye, Y. Li, L. Zhou, J. Liu and C. Wang, Green Chem., 2015, 17, 188–192 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1404849 and 1404844. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra23393c |
| ‡ Due to many chiral centres in our products, some products have more than two diastereoisomers. However, the mixture of the diastereomers couldn't be separated by column chromatography. Therefore, we selected the two major diastereoisomers to illustrate the phenomenon and one representative diastereoisomer to identify the structure of products in this work. |
| This journal is © The Royal Society of Chemistry 2016 |