
E,Z-Stereodivergent synthesis of N-tosyl α,β-dehydroamino esters via a Mukaiyama–Michael addition†
Miguel Espinosaa,
Andrea García-Ortiza,
Gonzalo Blay*a,
Luz Cardonaa,
M. Carmen Muñozb and
José R. Pedro*a
aDepartament de Química Orgànica, Facultat de Química, Universitat de València, C/Dr Moliner 50, E-46100 Burjassot, València, Spain. E-mail: jose.r.pedro@uv.es; gonzalo.blay@uv.es
bDepartament de Física Aplicada, Universitat Politècnica de València, Camí de Vera s/n, 46022 València, Spain
First published on 1st February 2016
Abstract
The stereodivergent synthesis of N-tosyl α,β-dehydroamino esters via a Mukaiyama–Michael addition is reported. The reaction of silylketene acetals with N-tosylimines derived from β,γ-unsaturated α-keto esters in dichloromethane provided the corresponding (Z)-α,β-dehydroamino esters while the (E)-isomers were obtained when the reaction was carried out in the presence of 10 mol% copper(II) triflate.
α,β-Dehydroamino acid derivatives are non-proteinogenic amino acids that are often found as structural subunits in natural products produced by bacteria, fungi, marine organisms and plants, and play an important role in the biosynthesis of other non-proteinogenic amino acids and D-amino acids (Fig. 1).1 Some of these compounds have shown antibiotic and other intriguing biological activities.2 The presence of the double bond in the dehydroamino acid residue reduces the conformational flexibility of peptides, a property that is useful for structure–activity studies and for the design of secondary structure in peptides,3 and it also confers resistance to enzymatic degradation and alters their bioactivity.4 These properties are affected by the E/Z configuration of the double bond of the dehydroamino acid moiety.5 Furthermore, α,β-dehydroamino acid derivatives are widely used as starting materials in the synthesis of natural and unnatural α-amino acids through catalytic hydrogenation,6 or conjugate addition,7 as well as in the synthesis of heterocyclic compounds.8 According to these pharmacological and synthetic potential, much synthetic effort has been devoted to the preparation of dehydroamino acids and their derivatives. Literature antecedents include elimination reactions of β-hydroxy-α-amino acids,9 Horner–Wadsworth–Emmons and Wittig reactions,10 condensation of aldehydes with N-protected glycine or 5-(4H)-oxazolone (Erlenmeyer synthesis),11 condensation of carbonyl compounds with isocyano acetates (Schöllkopf method),12 aminohalogenation of unsaturated esters followed by basic elimination,13 addition of amines to alkynoates,14 and Heck reaction.15 In most of these procedures the thermodynamically stable Z-isomer is predominantly formed,9a,10b,c,11,12b,c,13,14 while the synthesis of the E-isomer normally takes place with lower selectivity9b,10a,12a or requires the use of stereoisomerically pure starting materials that are usually prepared in multistep sequences or involves difficult isomer separation.9c,d
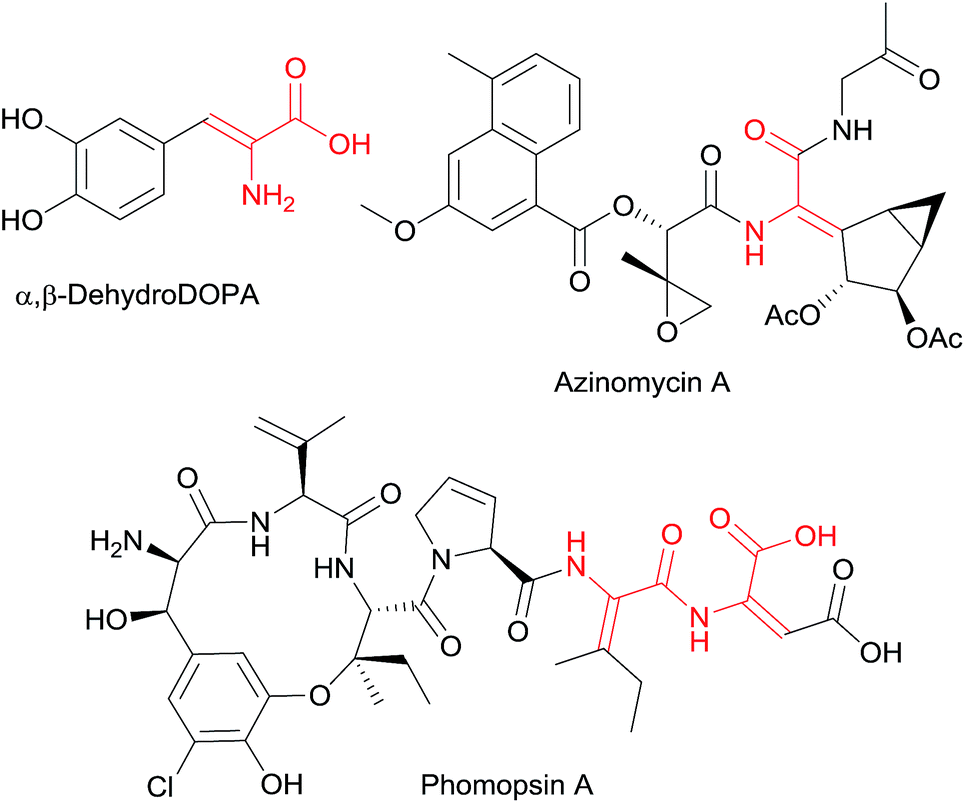 | ||
| Fig. 1 Examples of natural and bioactive α,β-dehydroamino acid derivatives. | ||
Recently, Palacios described a new approach to α,β-dehydroamino acid esters consisting in the conjugate addition of dialkyl zinc reagents to imines derived from β,γ-unsaturated α-keto esters catalysed by a copper(I)–phosphoramidite complex.16 A similar strategy has been reported by Liu and Hu for the construction of α-hydroxy-δ-amino esters from α-diazo esters and N-tosyl imines of β,γ-unsaturated α-keto esters17 and by Kim in the rhodium-catalysed 1,4 addition of arylboronic acids to unsaturated imino esters.18 In all these cases the reaction takes place stereoselectively to give the corresponding dehydroamino esters with the Z-configuration at the double bond. However, a procedure based on this approach that led stereoselectively to the E or Z dehydroamino esters starting from a same set of reactants has not been reported to date.19
Following our interest in nucleophilic addition reactions to unsaturated imines,20 we report herein our preliminary results in the E/Z stereodivergent synthesis of α,β-dehydroamino esters via a Mukaiyama–Michael addition of silylketene acetals to N-tosyl imines of β,γ-unsaturated α-keto esters (Scheme 1).
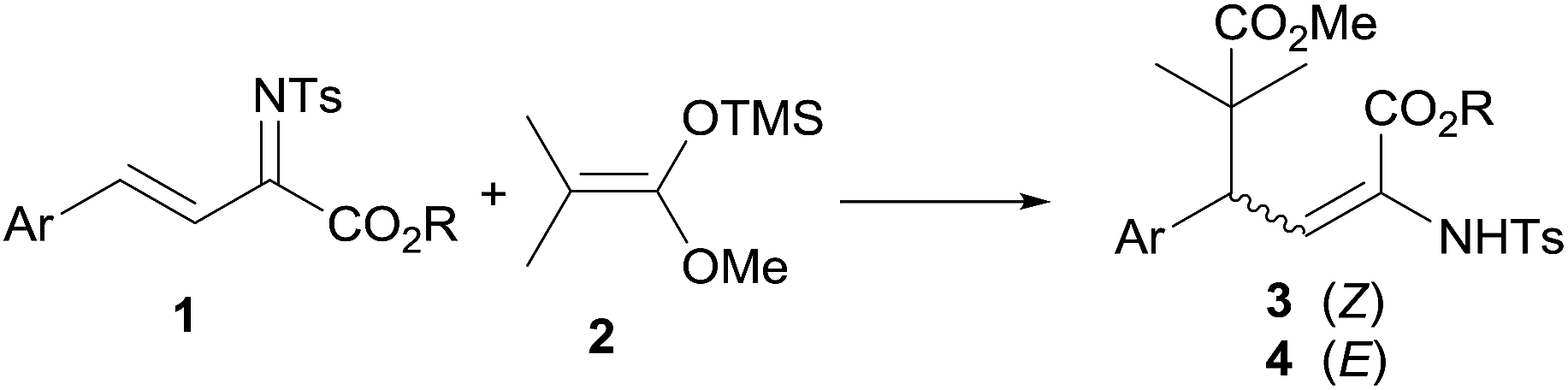 | ||
| Scheme 1 Mukaiyama–Michael reaction with N-tosyl imines of β,γ-unsaturated α-keto esters. | ||
The reaction between 2-methyl-1-methoxy-1-trimethylsilyoxyprop-1-ene (2) and imine 1a (Ar =Ph, R = Et) was carried out at room temperature.‡ The conjugate addition reaction proceeded smoothly in absence of any additive to give 85% yield of a mixture of two diastereomeric compounds 3a and 4a, differing in the geometry of the enamine double bond favouring the (Z)-isomer in a 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio (Table 1, entry 1). The reaction conditions were applied to a number of N-tosyl imines derived from ethyl γ-aryl-β,γ-unsaturated α-keto esters 1b–k, bearing either electron-donating or electron-withdrawing groups at the ortho, meta or para positions of the phenyl ring, and to an heteroaryl-substituted ester 1l. The reaction provided the expected α,β-dehydroamino esters favouring the formation of the (Z)-isomers 3b–l in all the examples studied, with diastereomeric ratios ranging from 85:15 to 99:1 (Table 1, entries 2–12). The methyl and isopropyl esters 1m and 1n gave similar results to ethyl ester 1a (Table 1, entries 12–13).
:9 ratio (Table 1, entry 1). The reaction conditions were applied to a number of N-tosyl imines derived from ethyl γ-aryl-β,γ-unsaturated α-keto esters 1b–k, bearing either electron-donating or electron-withdrawing groups at the ortho, meta or para positions of the phenyl ring, and to an heteroaryl-substituted ester 1l. The reaction provided the expected α,β-dehydroamino esters favouring the formation of the (Z)-isomers 3b–l in all the examples studied, with diastereomeric ratios ranging from 85:15 to 99:1 (Table 1, entries 2–12). The methyl and isopropyl esters 1m and 1n gave similar results to ethyl ester 1a (Table 1, entries 12–13).
| Entry | 1 | Ar | R | Yieldb 3 + 4 (%) | 3:4c (dr) |
|---|---|---|---|---|---|
| a 1 (0.25 mmol), 2 (0.6 mmol), CH2Cl2 (1.2 mL), rt, 20–24 h.b Yield of the diastereomer mixture after chromatography.c Determined by 1H NMR. | |||||
| 1 | 1a | Ph | Et | 84 | 91:9 |
| 2 | 1b | 4-MeC6H4 | Et | 77 | 91:9 |
| 3 | 1c | 4-MeOC6H4 | Et | 56 | 86:14 |
| 4 | 1d | 3-MeOC6H4 | Et | 82 | 89:11 |
| 5 | 1e | 2-MeOC6H4 | Et | 72 | 85:15 |
| 6 | 1f | 4-ClC6H4 | Et | 80 | 91:9 |
| 7 | 1g | 3-ClC6H4 | Et | 87 | 89:11 |
| 8 | 1h | 2-ClC6H4 | Et | 95 | 87:13 |
| 9 | 1i | 4-NO2C6H4 | Et | 98 | 92:8 |
| 10 | 1j | 3-NO2C6H4 | Et | 99 | 90:10 |
| 11 | 1k | 2-NO2C6H4 | Et | 58 | 99:1 |
| 12 | 1l | 2-Furyl | Et | 69 | 86:14 |
| 13 | 1m | Ph | Me | 82 | 89:11 |
| 14 | 1n | Ph | iPr | 75 | 89:11 |
On the other hand when the reaction between silyl ketene acetal 2 and imines 1a–n was carried out in the presence of 10 mol% copper(II) triflate in dichloromethane at room temperature,§ a faster reaction took place to give the expected α,β-dehydroamino esters in high yield, favouring in these cases the formation of the (E)-isomers 4b–n in all the examples studied (Table 2). In general, better yields and diastereoselectivities were observed for the copper-catalysed reaction compared with the non-catalysed reaction, with diastereomeric ratios (Z:E) above 10:90 in most of the cases.
| Entry | 1 | Ar | R | Yieldb 3 + 4 (%) | 3:4c (dr) |
|---|---|---|---|---|---|
| a 1 (0.25 mmol), 2 (0.6 mmol), Cu(OTf)2 (0.025 mmol), CH2Cl2 (1.9 mL), rt, 20–90 min.b Yield of the diastereomer mixture after chromatography.c Determined by 1H NMR. | |||||
| 1 | 1a | Ph | Et | 95 | 3:97 |
| 2 | 1b | 4-MeC6H4 | Et | 99 | 7:93 |
| 3 | 1c | 4-MeOC6H4 | Et | 95 | 12:88 |
| 4 | 1d | 3-MeOC6H4 | Et | 99 | 4:96 |
| 5 | 1e | 2-MeOC6H4 | Et | 91 | 3:97 |
| 6 | 1f | 4-ClC6H4 | Et | 99 | 6:94 |
| 7 | 1g | 3-ClC6H4 | Et | 99 | 3:97 |
| 8 | 1h | 2-ClC6H4 | Et | 99 | 1:99 |
| 9 | 1i | 4-NO2C6H4 | Et | 99 | 8:92 |
| 10 | 1j | 3-NO2C6H4 | Et | 99 | 6:94 |
| 11 | 1k | 2-NO2C6H4 | Et | 99 | 14:86 |
| 12 | 1l | 2-Furyl | Et | 80 | 9:91 |
| 13 | 1m | Ph | Me | 99 | 3:97 |
| 14 | 1n | Ph | iPr | 87 | 4:96 |
Initial attempts to assign the Z/E configuration of the double bond in compounds 3 and 4 by 1H NMR following spectroscopic criteria established by Mazurkiewicz et al.21 for related N-acyl-α,β-dehydro-α-amino acid esters led to contradictory results (see ESI†). NOESY experiments were then carried out with samples of products 3i and 4i¶ obtained under non-catalytic conditions and copper catalysis, respectively. NOESY experiments with compound 3i showed a small interaction between the NH proton (δ 6.16 ppm) and the benzylic proton (δ 4.69 ppm), which indicated the possible Z-geometry for the double bond in this compound. No significative interactions were found however in NOESY experiments with the Cu-catalysed reaction product 4i. On the other hand, suitable crystals for X-ray analysis of compound 4m (copper-catalysed reaction) could be obtained which allowed us to establish the (E)-configuration for the double bond in this compound (Fig. 2).|| On the basis of these results and 1H NMR characteristic chemical shifts (Table 3) we established that all compounds 3 resulting from the non-catalysed reaction have the (Z)-configuration at the double bond while this has the (E)-configuration for the copper-catalysed reaction products 4. Thus in all the cases the olefinic hydrogens appear at higher field for the E-isomer (δZCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) > δECH) while the N–H proton appears at higher field for the Z-isomer (δENH > δZNH). Similarly, the benzylic proton in the β-substituent also appears at higher field for the Z-isomer (δEArCH > δZArCH).
> δECH) while the N–H proton appears at higher field for the Z-isomer (δENH > δZNH). Similarly, the benzylic proton in the β-substituent also appears at higher field for the Z-isomer (δEArCH > δZArCH).
 | ||
| Fig. 2 Ortep plot for the X-ray structure of compound (E)-4m (copper catalysed reaction). The thermal ellipsoids are drawn at the 50% probability level. | ||
| Entry | 3,4 | Ar | R | δNH (Z)-3 | δNH (E)-4 | δCHC (Z)-3 |
δCHC (E)-4 |
δArCH (Z)-3 | δArCH (E)-4 |
|---|---|---|---|---|---|---|---|---|---|
| a 1H NMR carried out in CDCl3 at 300 MHz. δ values referenced to residual CHCl3 (δ = 7.26 ppm). | |||||||||
| 1 | a | Ph | Et | 6.20 | 6.61 | 7.31 | 7.01 | 4.41 | 4.81 |
| 2 | b | 4-MeC6H4 | Et | 6.14 | 6.58 | 7.29 | 6.97 | 4.35 | 4.77 |
| 3 | c | 4-MeOC6H4 | Et | 6.18 | 6.58 | 7.27 | 6.96 | 4.35 | 4.76 |
| 4 | d | 3-MeOC6H4 | Et | 6.18 | 6.59 | 7.27 | 6.97 | 4.37 | 4.79 |
| 5 | e | 2-MeOC6H4 | Et | 6.19 | 6.57 | 7.34 | 7.06 | 4.69 | 5.18 |
| 6 | f | 4-ClC6H4 | Et | 6.28 | 6.62 | 7.26 | 6.91 | 4.46 | 4.77 |
| 7 | g | 3-Cl-C6H4 | Et | 6.30 | 6.67 | 7.23 | 6.89 | 4.41 | 4.75 |
| 8 | h | 2-Cl-C6H4 | Et | 6.11 | 6.65 | 7.35 | 6.93 | 5.03 | 5.35 |
| 9 | i | 4-NO2C6H4 | Et | 6.16 | 6.71 | 7.31 | 6.90 | 4.69 | 4.88 |
| 10 | j | 3-NO2C6H4 | Et | 6.21 | 6.74 | 7.29 | 6.89 | 4.65 | 4.88 |
| 11 | k | 2-NO2C6H4 | Et | 6.65 | 6.69 | 7.03 | 6.88 | 4.93 | 5.42 |
| 12 | l | 2-Furyl | Et | 6.40 | 6.71 | 7.04 | 6.69 | 4.56 | 5.04 |
| 13 | m | Ph | Me | 6.23 | 6.57 | 7.30 | 7.00 | 4.35 | 4.72 |
| 14 | n | Ph | iPr | 6.24 | 6.60 | 7.29 | 6.98 | 4.45 | 4.86 |
The results indicate that the unsaturated imine 1 adopts the s-cis conformation in the non-catalytic reaction while it prefers the s-trans conformation during the copper-catalysed reaction. Although a full explanation for this preference is not possible at this moment, our working hypothesis is that the non-catalysed reaction takes place through a cyclic Diels–Alder-like transition state (Fig. 3, TS-1) with the incipient negative charge on the nitrogen atom stabilizing the incipient positive charge at the acetal carbon, which is only possible in the s-cis conformation.22 On the other hand, in the presence of copper triflate, coordination of the copper ion to the imine renders the substrate more electrophilic and the reaction takes place through an acyclic transition state (Fig. 3, TS-2) with the unsaturated imine having the s-trans conformation.**
 | ||
| Fig. 3 TS for the Mukaiyama–Michael reaction. Non-catalysed (TS-1) and copper-catalysed (TS-2). | ||
We are currently performing research addressed to study the scope of the reaction with other nucleophiles and mechanistic and theoretical studies which will be published conveniently.
Conclusions
We have developed a new procedure for the stereodivergent synthesis of N-tosyl α,β-dehydroamino esters via a Mukaiyama–Michael addition. The reaction of silylketene acetals with N-tosylimines derived from β,γ-unsaturated α-keto esters provided the corresponding (Z)-α,β-dehydroamino esters. On the other hand, the (E)-isomers were obtained when the reaction was performed in the presence of a catalytic amount of copper triflate. This allows the stereoselective synthesis of (E)- or (Z)-α,β-dehydroamino esters from a same set of reactants, which has no precedents in the literature. We have also established a correlation between the 1H NMR chemical shifts and the stereochemistry of the double bond in these compounds.Acknowledgements
Financial support from the MINECO (Spanish Government) and FEDER (EU) (CTQ2013-47494-P) and from Generalitat Valenciana (ISIC2012/001) is gratefully acknowledged. M. E. thanks the GV for a predoctoral grant. Access to NMR and MS facilities from the Servei Central de Suport a la Investigació Experimental (SCIE)-UV is also acknowledged.Notes and references
- (a) U. Kazmaier, Synthesis and Chemistry of α,β-Didehydroamino Acids, in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Andrew, Wiley-VCH, Weinheim, Germany, 2009, vol. 2, p. 3 Search PubMed; (b) F. S. Nandel and T. R. Sahrawat, Biopolymers, 2009, 92, 44 CrossRef CAS PubMed; (c) C. E. Ballard, H. Yu and B. Wang, Curr. Med. Chem., 2002, 9, 471 CrossRef CAS PubMed; (d) F. Sarabia, S. Chammaa, A. S. Ruiz, L. M. Ortiz and F. J. L. Herrera, Curr. Med. Chem., 2004, 11, 1309 CrossRef CAS PubMed; (e) T. V. Rajan Babu, Y. Y. Yan and S. Shin, Curr. Org. Chem., 2003, 7, 1759 CrossRef CAS; (f) C. Bonauer, T. Walenzky and B. Konig, Synthesis, 2006, 1 CAS; (g) J. Jiang, Z. Ma and S. L. Castle, Tetrahedron, 2015, 71, 5431 CrossRef CAS.
- (a) S. Wang, S. Zhou and W. Liu, Curr. Opin. Chem. Biol., 2013, 17, 626 CrossRef CAS PubMed; (b) M. Gupta and V. S. Chauhan, Biopolymers, 2011, 95, 161 CrossRef CAS PubMed; (c) B. Li, L. E. Cooper and W. A. van der Donk, Methods Enzymol., 2009, 458, 533 CAS; (d) P. M. T. Ferreira, L. S. Monteiro, T. Coban and S. Suzen, J. Enzyme Inhib. Med. Chem., 2009, 24, 967 CrossRef CAS PubMed; (e) R. Liao, L. Duan, L. Lian, P. Chun, H. Pan, Y. Ding, Q. Zhang, D. Chen, B. Shen, Y. Yu and W. Liu, Chem. Biol., 2009, 16, 141 CrossRef CAS PubMed.
- (a) M. Gupta and V. S. Chauhan, Biopolymers, 2011, 95, 161 CrossRef CAS PubMed; (b) D. Siodlak, M. Bujak and M. Stas, J. Mol. Struct., 2013, 1047, 229 CrossRef CAS; (c) J. S. Davies, J. Pept. Sci., 2003, 9, 471 CrossRef CAS PubMed; (d) A. S. Abreu, P. M. T. Ferreira, L. S. Monteiro, M. J. R. P. Queiroz, I. C. F. R. Ferreira, R. C. Calhelha and L. M. Estevinho, Tetrahedron, 2004, 60, 11821 CrossRef CAS; (e) C. Cativiela, M. D. Diaz de Villegas, J. A. Galvez and G. Su, ARKIVOC, 2004, 59 CAS.
- (a) P. Mathur, S. Ramakumar and V. S. Chauhan, Biopolymers, 2004, 76, 150 CrossRef CAS PubMed; (b) J. C. Jimenez, N. Bayo, B. Chavarria, A. Lopez-Macia, M. Royo, E. Nicolas, E. Giralt and F. Albericio, Lett. Pept. Sci., 2002, 9, 135 CAS.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475 CrossRef CAS PubMed.
- (a) M. A. Blaskovich, in Handbook on Syntheses of Amino Acids. General Route to Amino Acids, American Chemical Society and Oxford University Press, New York, 2010, p. 225 Search PubMed; (b) L. Panella, A. Marco-Aleixandre, G. J. Kruidhof, J. Robertus, B. L. Feringa, J. De Vries and A. J. Minnaard, J. Org. Chem., 2006, 71, 2026 CrossRef CAS PubMed; (c) D. J. Frank, A. Franzke and A. Pfaltz, Chem.–Eur. J., 2013, 19, 2405 CrossRef CAS PubMed.
- Z.-T. He, Y.-S. Zhao, P. Tian, C.-C. Wang, H.-Q. Dong and G.-Q. Lin, Org. Lett., 2014, 16, 1426 CrossRef CAS PubMed.
- (a) Z. Wang, M. Wang, X. Yao, Y. Li, J. Tan, L. Wang, W. Qiao, Y. Geng, Y. Liu and Q. Wang, Eur. J. Med. Chem., 2012, 54, 33 CrossRef CAS PubMed; (b) A. J. Ruger, M. Nieger and S. Brase, Tetrahedron, 2012, 68, 8823 CrossRef; (c) P. M. T. Ferreira, L. S. Monteiro and G. Pereira, Eur. J. Org. Chem., 2008, 4676 CrossRef CAS.
- (a) R. Ramesh, K. De and S. Chandrasekaran, Tetrahedron, 2007, 63, 10534 CrossRef CAS; (b) K. Godall and A. F. Parsons, Tetrahedron Lett., 1995, 3259 CrossRef; (c) L. Somekh and A. Shanzer, J. Org. Chem., 1983, 48, 907 CrossRef CAS; (d) F. Yokokawa and T. Shioiri, Tetrahedron Lett., 2002, 43, 8679 CrossRef CAS; (e) H. Sai, T. Ogiku and H. Omizu, Synthesis, 2003, 201 CrossRef CAS; (f) K. Nakamura, T. Isaka, H. Toshima and M. Kodaka, Tetrahedron Lett., 2004, 45, 7221 CrossRef CAS.
- (a) Y. Yasuno, M. Hamada, T. Yamada, T. Shinada and Y. Ohfune, Eur. J. Org. Chem., 2013, 1884 CrossRef CAS; (b) A. Baron, J. Martinez and F. Lamaty, Tetrahedron Lett., 2010, 51, 6246 CrossRef CAS; (c) U. Schmidt, H. Griesser, V. Leitenberger, A. Lieberknecht, R. Mangold, R. Meyer and B. Riedl, Synthesis, 1992, 487 CrossRef CAS; (d) U. Schmidt, A. Lieberknecht and J. Wild, Synthesis, 1984, 43 Search PubMed; (e) H. Azuma, K. Okuna, T. Fukuyama and H. Tokuyama, Org. Synth., 2011, 88, 152 CrossRef CAS.
- (a) T. Cleary, J. Brice, N. Kennedy and F. Chavez, Tetrahedron Lett., 2010, 51, 625 CrossRef CAS; (b) H. Zhao, S. G. Koenig, J. W. Dankwardt and S. P. Singh, Org. Process Res. Dev., 2014, 18, 198 CrossRef CAS; (c) B. S. Jursic, S. Sagiraju, D. K. Ancalde, T. Clark and E. D. Stevens, Synth. Commun., 2007, 37, 1709 CrossRef CAS.
- (a) D. Hoppe and U. Schöllkopf, Angew. Chem., Int. Ed., 1970, 9, 300 CrossRef CAS; (b) D. Enders, Z.-X. Chen and G. Raabe, Synthesis, 2005, 306 CrossRef CAS; (c) C. Benhaim, L. Bouchard, G. Pelletier, J. Sellstedt, L. Kristofova and S. Daigneault, Org. Lett., 2010, 12, 2008 CrossRef CAS PubMed.
- D. D. Chen, L. Guo, J. Liu, S. Kirtane, J. F. Cannon and G. Li, Org. Lett., 2005, 7, 921 CrossRef CAS PubMed.
- (a) B. M. Trost and G. R. Dake, J. Am. Chem. Soc., 1997, 119, 7595 CrossRef CAS; (b) G. Choudhary and R. K. Peddinti, Green Chem., 2011, 13, 3290 RSC; (c) A. S. Chavan, J.-C. Deng and S.-C. Chuang, Molecules, 2013, 18, 2611 CrossRef CAS PubMed.
- R. O. Cann, C.-P. H. Chen, Q. Gao, R. L. Hanson, D. Hsieh, J. Li, D. Lin, R. L. Parsons, Y. Pendri, R. B. Nielsen, W. A. Nugent, W. L. Parker, S. Quinlan, N. P. Reising, B. Remy, J. Sausker and X. Wang, Org. Process Res. Dev., 2012, 16, 1953 CrossRef CAS.
- (a) F. Palacios and J. Vicario, Synthesis, 2007, 3923 CrossRef CAS; (b) F. Palacios and J. Vicario, Org. Lett., 2006, 8, 5405 CrossRef CAS PubMed.
- L. Qiu, L. Gao, J. Tang, D. Wang, X. Guo, S. Liu, L. Yang, J. Li and W. Hu, J. Org. Chem., 2014, 79, 4142 CrossRef CAS PubMed.
- A. Lee and H. Kim, J. Am. Chem. Soc., 2015, 137, 11250 CrossRef CAS PubMed.
- Cyclic derivatives of dehydroamino acids have been prepared from β,γ-unsaturated α-imino esters through different cyclisation strategies: (a) F. Palacios, J. Vicario and D. Aparicio, Eur. J. Org. Chem., 2006, 2843 CrossRef CAS; (b) S.-L. Zhou, J.-L. Li, L. Dong and Y.-C. Chen, Org. Lett., 2011, 13, 5874 CrossRef CAS PubMed; (c) J. Tian and Z. He, Chem. Commun., 2013, 49, 2058 RSC; (d) Y. Deng, L. Liu, R. G. Sarkisian, K. Wheeler, H. Wang and Z. Xu, Angew. Chem., Int. Ed., 2013, 52, 3663 CrossRef CAS PubMed.
- (a) M. Espinosa, G. Blay, L. Cardona and J. R. Pedro, Chem.–Eur. J., 2013, 19, 14861 CrossRef CAS PubMed; (b) M. Espinosa, G. Blay, L. Cardona and J. R. Pedro, Chem.–Eur. J., 2013, 19, 17632 CrossRef CAS.
- R. Mazurkiewicz, A. Kuznik, M. Grymel and N. Kuznik, Magn. Reson. Chem., 2005, 43, 36 CrossRef CAS PubMed.
- (a) D. L. Boger and W. L. Corbett, J. Org. Chem., 1993, 58, 2068 CrossRef CAS; (b) J. Vicario, D. Aparicio and F. Palacios, Tetrahedron Lett., 2011, 52, 4109 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures. Copies of NMR spectra for new compounds. X-ray structures for compounds 1b and 4m. CCDC 1442362 and 1442206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra27354d |
| ‡ 2-Methyl-1-methoxy-1-trimethylsilyoxyprop-1-ene (2, 122 μL, 0.6 mmol) was added to a solution of imine 1 (0.25 mmol) in CH2Cl2 (1.2 mL) at rt under nitrogen atmosphere and the mixture was stirred overnight until the reaction was complete (20–24 h). The reaction products were isolated by column chromatography on silica gel eluting with hexane/EtOAc mixtures. |
| § A solution of imine 1 (0.25 mmol) in dry CH2Cl2 (1 mL) was added to a suspension of anhydrous Cu(OTf)2 (9 mg, 0.025 mmol) in dry CH2Cl2 (0.9 mL) contained in a Schlenck tube under nitrogen at rt, followed by 2-methyl-1-methoxy-1-trimethylsilyoxyprop-1-ene (2, 122 μL, 0.6 mmol). The mixture was stirred until the reaction was complete (30–90 min). The reaction products were isolated by column chromatography on silica gel eluting with hexane/EtOAc mixtures. |
| ¶ Compounds 3i and 4i, i.e. p-nitro derivatives, were used to avoid overlapping of 1H NMR signals in the aromatic region. |
| || CCDC 1442362. |
| ** X-ray analysis of imine 1b indicates the (Z)-geometry of the CN bond and the preference of the s-trans conformation in the crystal structure (see ESI). CCDC 1442206. |
| This journal is © The Royal Society of Chemistry 2016 |