
Theoretical and experimental investigations of the Li storage capacity in single-walled carbon nanotube bundles†
Abstract
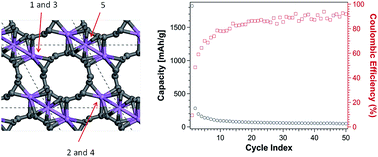
Single-walled carbon nanotube bundles are investigated as Li-ion battery anode materials using a theoretical and experimental approach. Density functional theory yields Li binding energies and the intercalation capacity of a fixed density bundle of identical tubes. Two nanotube diameters are tested to explore the size effect on Li storage capacity. An infinite bundle model represents low surface area and a bundle with open tubes describes high surface area tuned by edges terminated in low-coordinated sites or functionalized with hydrogen. Charge and discharge curves are measured during the first 5 cycles and the chemistry of intercalation and interfacial reactions are characterized via in situ Raman spectroscopy. A significantly high capacity at the first discharge featuring Li intercalation and products from interfacial reactions evident from Raman spectroscopy is dramatically reduced at the 2nd cycle and still decreases during successive cycles. The experimentally observed irreversible capacity loss is explained by the density functional theory analyses that demonstrate a much lower capacity of the nanotube bundle compared to graphite, in agreement with the results obtained in the 2nd cycle. The excess capacity measured in the first cycle is attributed to the presence of defects and unsaturated edges and to the formation of a solid-electrolyte interphase.
 Please wait while we load your content...
Please wait while we load your content...

