
Galvanic replacement of electrodeposited nickel by palladium and investigation of the electrocatalytic activity of synthesized Pd/(Ni) for hydrogen evolution and formic acid oxidation†
Abstract
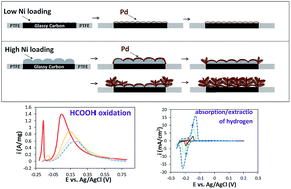
A series of controllable Pd/(Ni) catalysts were synthesized by the use of galvanic replacement between electrodeposited nickel and palladium in [PdCl4]2− containing solution. The mechanisms of galvanic replacement were investigated by monitoring the potential variations versus time, field emission scanning electron microscopy (FESEM), and energy dispersive X-ray spectroscopy (EDS). The electrocatalytic activities of the prepared catalysts were evaluated by linear sweep voltammetry for the hydrogen evolution reaction (HER) in 0.5 M H2SO4 and formic acid oxidation in 0.5 M H2SO4 + 0.5 M HCOOH. In addition, the effect of the Ni loading amount on the final morphology, chemical composition and electrocatalytic activity of the galvanically replaced Pd/(Ni) catalysts was studied. The results indicated that the morphology of the Pd/(Ni) changes from thin film to nano/micro dendrite depending on the amounts of electrodeposited Ni loading. The sharp and relatively symmetric peaks that were observed in the voltammograms indicated the reversibility of electrochemical hydrogen absorption/extraction. The same Tafel slopes estimated for all Pd/(Ni) suggested the predomination of Volmer–Heyrovsky mechanisms in catalyzing the hydrogen evolution reaction. In addition, the Pd/(Ni) thin film catalyst exhibited superior electrocatalytic mass activity towards the oxidation of formic acid (1.39 A mg−1 at the peak potential). Moreover, comparison of the Pd/(Ni) thin film activity with some other palladium-based catalysts in some recent studies with relatively good results showed that the galvanically replaced Pd/(Ni) thin film has great potential as a fast, easily prepared, and active electrocatalyst for formic acid oxidation.
 Please wait while we load your content...
Please wait while we load your content...

