
Differential influence of additives on the various stages of insulin aggregation†
Abstract
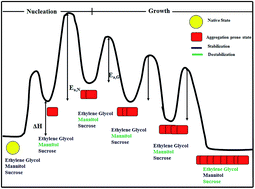
The kinetics of aggregation of insulin at pH 1.6 was studied by monitoring the turbidity as a function of time. The aggregation of insulin occurs by a typical nucleation-growth mechanism, resulting in a distinct lag phase, followed by a growth phase and a saturation phase. Sugars and polyols affect the lag time and the rate constant of growth to different extents, suggesting that these additives affect both the pre- and post-nucleation processes. Sucrose and ethylene glycol effectively reduce the aggregation, whereas mannitol is not effective in suppressing the overall aggregation. However, it can delay the aggregation process. It is also inferred that nucleation and elongation may not be affected in the same manner in the presence of a particular additive. The varying behaviour of different additives suggests their interference in the different stages of aggregation, affecting the aggregation intermediates differently. The viscosity of the medium has a minimal effect on the nucleation process, but does affect the elongation rate. The temperature dependence of the aggregation profiles provides probable reasons for the inhibitory action of these additives. The results also suggest the existence of different types of interactions between the additives and the various species in the aggregation pathway of insulin.
 Please wait while we load your content...
Please wait while we load your content...

