
Correlating the magnetism and gas sensing properties of Mn-doped ZnO films enhanced by UV irradiation†
Abstract
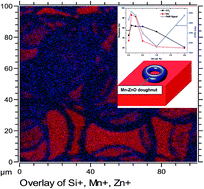
In this study, we report on the correlation between the magnetism and gas sensing properties of Mn-doped ZnO films grown via aerosol spray pyrolysis. The evolution of the structure, morphology, optical properties, and chemical state of ZnO with the Mn concentration was also investigated. ZnO doped with Mn (0.1 at%) demonstrated room-temperature ferromagnetism (RTFM) due to the uncompensated surface spins primarily originating from structural defects and oxygen vacancies (VO) on the surface, which act as active sites for the adsorption of oxygen species. The undoped ZnO structure revealed both FM and paramagnetism (PM) at the near surface of the film. Increased Mn doping destroyed the RTFM ordering due to improved PM features induced by Mn clusters on the ZnO surface and reduced amount of VO on the surface. However, ZnO films doped with Mn (0.1 at%) exhibited an improved sensing response to oxidizing gases compared to their counterparts, showing that films with RTFM with no PM contribution exhibit improved sensing properties. These analyses revealed that the nature of the film surface plays a substantial role in both their magnetic and sensing behaviors.
 Please wait while we load your content...
Please wait while we load your content...

