
Copper-catalysed synthesis of trifluoromethyl(hetero)arenes from di(hetero)aryl-λ3-iodanes†
Vinay Kumar Pandey and
Pazhamalai Anbarasan*
Department of Chemistry, Indian Institute of Technology Madras, Chennai-600036, India. E-mail: anbarasansp@iitm.ac.in
First published on 9th February 2016
Abstract
An efficient synthesis of trifluoromethylated (hetero)arenes has been achieved through the regioselective copper-catalyzed trifluoromethylation of di(hetero)aryl-λ3-iodanes, employing readily available trifluoromethyltrimethylsilane. The reaction works well for both symmetrical and unsymmetrical di(hetero)aryl-λ3-iodanes with good regioselectivity and also tolerates diverse functional groups such as bromo, iodo, cyano, nitro, ester, ketone and enolizable ketone.
Trifluoromethylated (hetero)arenes represent a prevalent subunit in agrochemicals, pharmaceuticals and advanced organic materials.1 The introduction of the trifluoromethyl (CF3) group into the potent organic molecules often drastically alters their physicochemical properties.2 Representative examples of trifluoromethylated arenes containing pharmaceutical drugs are shown in Fig. 1. However, trifluoromethylated compounds are not found in nature, despite the large abundance of fluorine in the earth's crust, because of which the development of practical and new methodologies for the introduction of the ‘CF3’ group has significantly attracted the attention of the scientific community in recent years.3
 | ||
| Fig. 1 Trifluoromethylated arene containing pharmaceutically important molecules. | ||
Traditionally, synthesis of trifluoromethylated arenes were achieved via the Swarts reaction of toluene derivatives, but the utility is rather limited due to the low functional group compatibility and toxicity of the reagent.4 In recent years, they were replaced with Cu/Pd mediated direct trifluoromethylation of prefunctionalized arenes5 such as aryl halides,6 arylboron reagents,7 (hetero)arenes8 using either nucleophilic trifluoromethylating reagent (Ruppert–Prakash reagent)9 or electrophilic trifluoromethylating reagent10 (Umemoto's11 and Togni's reagents12). Recently, aryldiazonium salts, an electrophilic arylating reagent were also trifluoromethylated employing super stoichiometric amount of Cu or Ag salts.13 In general, most of these reactions suffer from high-catalyst loading and harsh reaction conditions. Thus, mild and efficient processes for the introduction of trifluoromethyl group onto aromatic rings are rather highly desirable.
Diaryl-λ3-iodanes have emerged as an alternative and efficient electrophilic arylating agent in modern organic synthesis for the C–C and C-heteroatom bond forming transformations,14 since they are readily accessible from simple arenes. Very recently, Qing and co-workers reported the copper mediated trifluoromethylation of diaryl-λ3-iodanes.15 However, this method requires a stoichiometric amount of copper catalyst, thus there is a great in developing general and catalytic method for the regioselective trifluoromethylation of diaryl-λ3-iodanes.16 Owing to the potential of trifluoromethylated aromatic compounds and based on our continued interest in the synthesis of fluorinated organics and use of diaryl-λ3-iodanes,17 herein we reveal the elegant copper-catalyzed regioselective trifluoromethylation of diaryl-λ3-iodanes employing readily available trifluoromethyltrimethylsilane, TMSCF3 (Scheme 1).
 | ||
| Scheme 1 An approach to direct trifluoromethylation of di(hetero)aryl-λ3-iodanes. | ||
For our initial studies, we chose symmetrical diphenyl-λ3-iodane 1a as model substrate, which was synthesized from readily available benzene and iodine under oxidative conditions.18 The reaction of 1 equivalent of 1a and 2 equivalents of TMSCF3 in presence of 20 mol% of CuI in DMF at various temperature did not afford any detectable amount of trifluoromethylbenzene 2a, in 19F NMR (Table 1). To our delight, addition of 1 equivalent of CsF to release the CF3-anion from TMSCF3 promoted the expected reaction to furnish the product 2a in 50% yield, based on 19F NMR (Table 1, entry 2). Subsequently, screening of various additives revealed that KF is best promoter for the present trifluoromethylation with 75% yield (Table 1, entries 3–5). On the other hand, no trifluoromethylation of 1a with TMSCF3 and KF was observed in the absence CuI (Table 1, entry 6). This revealed that the present trifluoromethylation indeed was catalyzed by CuI and promoted by KF.
|
||||
|---|---|---|---|---|
| Entry | X (mol%) | Ligand (Y mol%) | Additive | Yieldb (%) |
| a Reaction conditions: 1a (50 mg, 0.115 mmol, 1 equiv.), TMSCF3 (0.23 mmol, 2 equiv.), CuI (X mol%), ligand (Y mol%), additive (1 equiv.), DMF, 65 °C, 12 h.b All are 19F NMR yields.c Cu(OTf)2 is used.d Cu(CH3CN)4BF4 is used.e At room temperature. | ||||
| 1 | 20 | — | — | 0 |
| 2 | 20 | — | CsF | 50 |
| 3 | 20 | — | KF | 75 |
| 4 | 20 | — | TBAF | 15 |
| 5 | 20 | — | tBuOK | 7 |
| 6 | — | — | KF | 0 |
| 7 | 10 | — | KF | 65 |
| 8 | 10 | PPh3 (10) | KF | 85 |
| 9 | 10 | P(o-Tolyl)3 (10) | KF | 88 |
| 10 | 10 | X-Phos (10) | KF | 91 |
| 11 | 10 | Bipy (10) | KF | 48 |
| 12 | 10 | Phen (10) | KF | 71 |
| 13 | 10c | PPh3 (10) | KF | 10 |
| 14 | 10d | PPh3 (10) | KF | 6 |
| 15e | 10 | PPh3 (10) | KF | 20 |
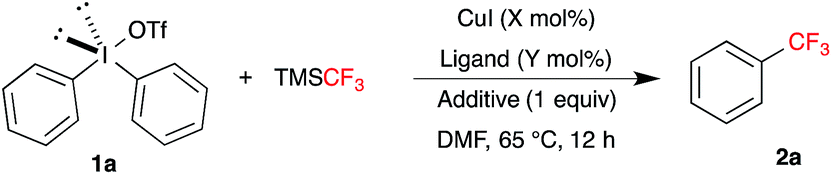
Next, decreasing the catalyst loading to 10 mol% gave the product 2a in low yield (65%, Table 1, entry 7). To improve the efficiency of the reaction with low catalyst loading, various phosphorous and nitrogen based ligands were examined. Reactions in the presence of electron rich phosphine ligands (PPh3, P(o-tolyl)3 and X-Phos) afforded the product 2a in excellent yield (Table 1, entries 8–10). In contrast, nitrogen based ligands did not improve the outcome of the present trifluoromethylation (Table 1, entries 11–12). Considering the efficiency and cost of phosphine ligands, PPh3 was chosen as suitable ligand for further investigation. Screening of other copper salts such as Cu(OTf)2 and Cu(CH3CN)4BF4 gave the product in very low yield (∼10%, Table 1, entries 13 and 14). Similar decrease in yield (20%) was observed when the temperature was reduced to room temperature (Table 1, entry 15). Finally, entry 8 in Table 1 was chosen as optimal reaction conditions for the further exploration of substrates scope.
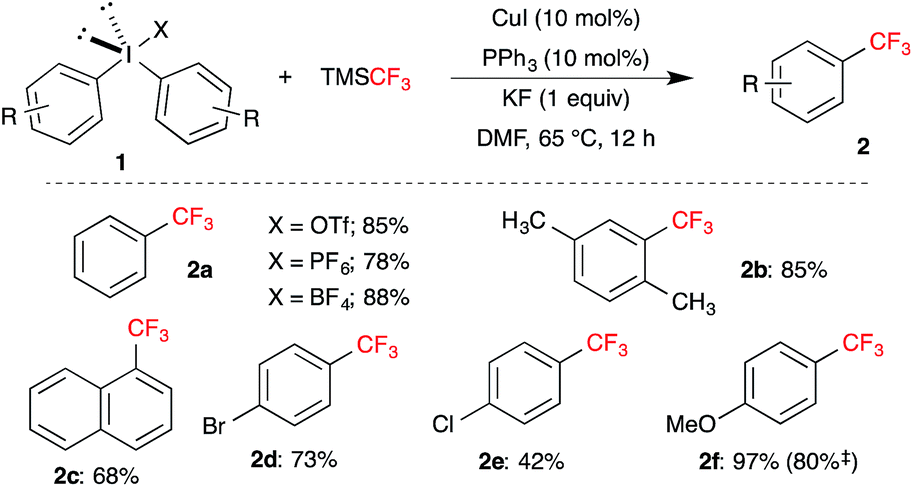
Having achieved the optimal reaction condition for the copper-catalyzed trifluoromethylation, scope and limitation of various symmetrical diaryl-λ3-iodanes 1 was investigated. As shown in Scheme 2, diverse substituted trifluoromethylbenzenes 2 were achieved in moderate to excellent yield. Initial screening of non-coordinating counterion like PF6-anion and BF4-anion gave the product 2a in comparable yield. Electron deficient halo-substituted trifluoromethylbenzene 2d and 2e were synthesized in good yield from corresponding symmetrical diaryl-λ3-iodanes. Similarly, electron rich methoxy substituted trifluoromethylbenzene 2f was achieved in 80% isolated yield. Interestingly, the present reaction conditions also tolerate the sterically demanding ortho-substituted diaryl-λ3-iodanes and led to the formation of 2b and 2c in 85% and 68% yield, respectively.
 | ||
| Scheme 2 Copper-catalyzed trifluoromethylation of symmetrical diaryl-λ3-iodanes. | ||
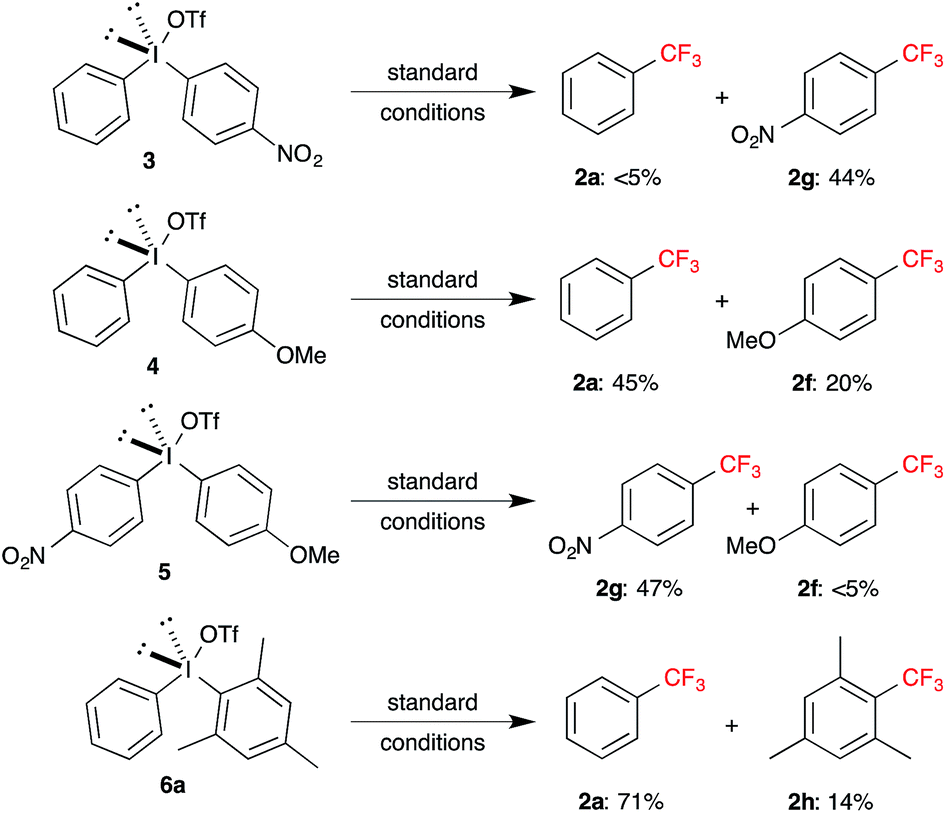
Although the developed trifluoromethylation of symmetrical diaryl-λ3-iodanes has shown significant scope, synthesis of highly electronically rich and deficient aryl containing symmetrical diaryl-λ3-iodanes are rather difficult.18 On the other hand, synthesis of unsymmetrical diaryl-λ3-iodanes is relatively easier, but the regioselective functionalization is a potential problem.19 Hence, we focused our attention on the trifluoromethylation of unsymmetrical diaryl-λ3-iodanes employing the optimized conditions. At first, we synthesized electronically and sterically different diaryl-λ3-iodanes (3–6) and subjected under the copper-catalyzed trifluoromethylation conditions. Reaction of diaryl-λ3-iodane 3 containing electronically neutral (Ph) and deficient (4-NO2Ph) aryl moiety afforded the trifluoromethylated product 2g as predominant product (Scheme 3). On the other hand, ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trifluoromethylated products 2a and 2f were observed with unsymmetrical diaryl-λ3-iodane 4, where electronically neutral aryl moiety was trifluoromethylated faster than electron rich aryl moiety. Next, trifluoromethylation of nitro and methoxy substituted aryl containing unsymmetrical diaryl-λ3-iodane 5 gave product 2g as major product. Interestingly, sterically different phenyl and mesityl substituted diaryl-λ3-iodane 6a afforded benzotrifluoride (2a) as major product along with minor amount of mesityl trifluoromethylated product 2h (Scheme 3). These results show that regioselective trifluoromethylation of unsymmetrical diaryl-λ3-iodanes mainly depends on the electronic and steric factor of aryl groups; specifically, trifluoromethylation is preferred at electron deficient and sterically less hindered aryl moiety.
:1 mixture of trifluoromethylated products 2a and 2f were observed with unsymmetrical diaryl-λ3-iodane 4, where electronically neutral aryl moiety was trifluoromethylated faster than electron rich aryl moiety. Next, trifluoromethylation of nitro and methoxy substituted aryl containing unsymmetrical diaryl-λ3-iodane 5 gave product 2g as major product. Interestingly, sterically different phenyl and mesityl substituted diaryl-λ3-iodane 6a afforded benzotrifluoride (2a) as major product along with minor amount of mesityl trifluoromethylated product 2h (Scheme 3). These results show that regioselective trifluoromethylation of unsymmetrical diaryl-λ3-iodanes mainly depends on the electronic and steric factor of aryl groups; specifically, trifluoromethylation is preferred at electron deficient and sterically less hindered aryl moiety.
 | ||
| Scheme 3 Regioselective trifluoromethylation of 3–6. | ||
After the successful study of regioselective trifluoromethylation of unsymmetrical diaryl-λ3-iodanes, we focused our attention on the generality of the regioselective trifluoromethylation of various unsymmetrical diaryl-λ3-iodanes having mesityl as constant blocker. Various electronically and sterically diverse unsymmetrical diaryl-λ3-iodanes having mesityl group were regioselectively trifluoromethylated in good to excellent yield under the optimized conditions, along with ∼5–10% of 2h (Scheme 4). Simple alkyl and aryl substituted phenyl containing unsymmetrical diaryl-λ3-iodanes furnished the expected products 2i–k, 2m and 2n in ∼60–84% yield, among them 2k was isolated in 70% yield. ortho-Trifluoromethyltoluene 2l was achieved in 65% yield from corresponding unsymmetrical diaryl-λ3-iodane. Readily functionalizable and reactive fluoro, bromo and iodo substituted unsymmetrical diaryl-λ3-iodanes were well tolerated under the optimized conditions to afford the corresponding benzotrifluorides (2o, 2d and 2p) in moderate to good yield. Interestingly, the present trifluoromethylation is highly compatible with reactive functional groups such as trifluoromethyl, nitrile, nitro, ester, ketone and enolizable ketone and led to the corresponding benzotrifluorides (2q–2v) in ∼55–94% yield, wherein 2s and 2t were isolated in 65% and 86% yield, respectively. Furthermore, thiophen-2-yl, 2-chloropyridin-5-yl and quinol-6-yl, heteroaromatic substituted unsymmetrical diaryl-λ3-iodanes were successfully trifluoromethylated in good yield and regioselectivity. These studies revealed that the present optimized condition is applicable for regioselective trifluoromethylation of both symmetrical and unsymmetrical diaryl-λ3-iodanes in high yield.
 | ||
| Scheme 4 Regioselective trifluoromethylation of unsymmetrical diaryl-λ3-iodanes. All are 19F NMR yields. In all the cases ∼5–10% of 2 h was observed. †∼20% of 2 h was observed. ‡Isolated yield. | ||
Next, preliminary mechanistic investigations were carried out on the optimized reaction. Since many of the copper-catalyzed transformations were proposed to involve radical species,13b,13c,20 influence of radical scavenger and radical clock probe experiments were envisioned to validate whether the radical species are formed in the present trifluoromethylation conditions.
Trifluoromethylation of 1a in the presence of excess of TEMPO under the standard conditions gave the product 2a in 89% yield (Scheme 5a), which is similar to the yield obtained in Table 1. The formation of either TEMPO-CF3 7 or TEMPO-Ph was not observed, as determined by 1H, 19F NMR and GCMS analysis of the crude reaction mixture. Even formation of 7 was not observed with TMSCF3 and TEMPO in the absence of 1a under the standard conditions.
 | ||
| Scheme 5 Mechanistic investigation. | ||
To rule out the formation of aryl radical species, allylether substituted diaryl-λ3-iodane 6z, radical clock probe, was subjected under the standard conditions. The trifluoromethylated product 2z was formed in 36% yield along with 50% of 2h, whereas cyclization product 8 was not observed, as determined by NMR and GCMS (Scheme 5b). The increased yield of 2h and diminished yield of 2z were possibly due to the high electron rich nature of allyloxyphenyl moiety. These observations are not in support of the generation of radical species in the present reaction conditions. Next, to probe insights into the mechanism of copper-catalyzed trifluoromethylation, 31P NMR and 19F NMR studies were conducted. Unfortunately, all these studies did not afford any valuable information about the mechanism (see ESI†).
Thus, based on the observation and copper-catalyzed cross-coupling reaction,20,21 we postulated the following mechanism for the copper-catalyzed trifluoromethylation of symmetrical and unsymmetrical diaryl-λ3-iodanes 1/6 (Scheme 6). The active copper complex A, generated from Cu(I) and PPh3, on reaction with TMSCF3 in the presence of KF would form the CF3-bound copper species B. Formation of copper species C could be explained through the oxidation of B with diaryl-λ3-iodanes 1/6. Finally, reductive elimination of benzotrifluorides 2 from C would afford the active copper species A to continue the catalytic cycle.
 | ||
| Scheme 6 Plausible mechanism. | ||
In conclusion, an efficient copper-catalyzed trifluoromethylation of symmetrical and unsymmetrical diaryl-λ3-iodanes have been developed for the synthesis of diverse trifluoromethylated (hetero)arenes in good yield and regioselectivity. The optimized reaction tolerates various reactive functional groups like bromo, iodo, cyano, nitro, ester, ketone and enolizable ketone.
Acknowledgements
We thank Council of Scientific & Industrial Research (CSIR) (Project No. 02(0092)/12/EMR-II) for financial support and Department of Science and Technology (DST) (Project No. SR/S1/OC-48/2012) for sponsoring Gas Chromatography. VKP thanks CSIR for a fellowship.Notes and references
- (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH Verlag GmbH, Weinheim, 2013 Search PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- (a) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (b) G. K. Prakash and M. Mandal, J. Fluorine Chem., 2001, 112, 123 CrossRef CAS; (c) J.-A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975 CrossRef CAS; (d) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS PubMed; (e) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (f) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed.
- J. H. Simons and C. J. Lewis, J. Am. Chem. Soc., 1938, 60, 492 CrossRef CAS.
- (a) P. Chen and G. Liu, Synthesis, 2013, 45, 2919 CrossRef CAS; (b) X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 1744 CrossRef CAS PubMed; (c) S. Roy, B. T. Gregg, G. W. Gribble, V.-D. Le and S. Roy, Tetrahedron, 2011, 67, 2161 CrossRef CAS.
- (a) A. Maleckis and M. S. Sanford, Organometallics, 2014, 33, 2653 CrossRef CAS; (b) X. Li, J. Zhao, L. Zhang, M. Hu, L. Wang and J. Hu, Org. Lett., 2015, 17, 298 CrossRef CAS PubMed; (c) T. Ruhl, W. Rafique, V. T. Lien and P. J. Riss, Chem. Commun., 2014, 50, 6056 RSC; (d) M. G. Mormino, P. S. Fier and J. F. Hartwig, Org. Lett., 2014, 16, 1744 CrossRef CAS PubMed; (e) A. Lishchynskyi, M. A. Novikov, E. Martin, E. C. Escudero-Adan, P. Novak and V. V. Grushin, J. Org. Chem., 2013, 78, 11126 CrossRef CAS PubMed; (f) M. Huiban, M. Tredwell, S. Mizuta, Z. Wan, X. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941 CrossRef CAS PubMed; (g) H. Kondo, M. Oishi, K. Fujikawa and H. Amii, Adv. Synth. Catal., 2011, 353, 1247 CrossRef CAS; (h) T. Knauber, F. Arikan, G.-V. Roschenthaler and L. J. Gooßen, Chem.–Eur. J., 2011, 17, 2689 CrossRef CAS PubMed; (i) M. Oishi, H. Kondo and H. Amii, Chem. Commun., 2009, 1909 RSC; (j) Z. Gonda, S. Kovacs, C. Weber, T. Gati, A. Meszaros, A. Kotschy and Z. Novak, Org. Lett., 2014, 16, 4268 CrossRef CAS PubMed; (k) Z. Weng, R. Lee, W. Jia, Y. Yuan, W. Wang, X. Feng and K.-W. Huang, Organometallics, 2011, 30, 3229 CrossRef CAS.
- (a) S. Arimori and N. Shibata, Org. Lett., 2015, 17, 1632 CrossRef CAS PubMed; (b) P. Ivashkin, G. Lemonnier, J. Cousin, V. Gregoire, D. Labar, P. Jubault and X. Pannecoucke, Chem.–Eur. J., 2014, 20, 9514 CrossRef CAS PubMed; (c) H. Serizawa, K. Aikawa and K. Mikami, Org. Lett., 2014, 16, 3456 CrossRef CAS PubMed; (d) S. R. Dubbaka, M. Salla, R. Bolisetti and S. Nizalapur, RSC Adv., 2014, 4, 6496 RSC; (e) M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 12837 CrossRef CAS PubMed; (f) Y. Li, L. Wu, H. Neumann and M. Beller, Chem. Commun., 2013, 49, 2628 RSC; (g) P. Novak, A. Lishchynskyi and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 7767 CrossRef CAS PubMed; (h) B. A. Khan, A. E. Buba and L. J. Gooßüen, Chem.–Eur. J., 2012, 18, 1577 CrossRef CAS PubMed.
- (a) S. Zhong, A. Hafner, C. Hussal, M. Nieger and S. Brase, RSC Adv., 2015, 5, 6255 RSC; (b) M. Wu, X. Ji, W. Dai and S. Cao, J. Org. Chem., 2014, 79, 8984 CrossRef CAS PubMed; (c) J. Xie, X. Yuan, A. Abdukader, C. Zhu and J. Ma, Org. Lett., 2014, 16, 1768 CrossRef CAS PubMed; (d) F. Sladojevich, E. McNeill, J. Borgel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712 CrossRef CAS PubMed; (e) G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38 CrossRef CAS PubMed; (f) S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2013, 49, 6385 RSC; (g) L.-S. Zhang, K. Chen, G. Chen, B.-J. Li, S. Luo, Q.-Y. Guo, J.-B. Wei and Z.-J. Shi, Org. Lett., 2013, 15, 10 CrossRef CAS PubMed; (h) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- (a) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (b) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- S. Barata-Vallejo, B. Lantano and A. Postigo, Chem.–Eur. J., 2014, 20, 16806 CrossRef CAS PubMed.
- C. Zhang, Org. Biomol. Chem., 2014, 12, 6580 CAS.
- J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- (a) D. L. Browne, Angew. Chem., Int. Ed., 2014, 53, 1482 CrossRef CAS PubMed; (b) A. Lishchynskyi, G. Berthon and V. V. Grushin, Chem. Commun., 2014, 50, 10237 RSC; (c) G. Danoun, B. Bayarmagnai, M. F. Grunberg and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 7972 CrossRef CAS PubMed; (d) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330 CrossRef CAS PubMed.
- (a) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (b) S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS PubMed; (c) L. Chan, A. McNally, Q. Y. Toh, A. Mendoza and M. J. Gaunt, Chem. Sci., 2015, 6, 1277 RSC; (d) I. Sokolovs, D. Lubriks and E. Suna, J. Am. Chem. Soc., 2014, 136, 6920 CrossRef CAS PubMed.
- J.-Y. Yang, X.-H. Xu and F.-L. Qing, J. Fluorine Chem., 2015, 180, 175 CrossRef CAS.
- J. R. Bour, N. M. Camasso and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 8034 CrossRef CAS PubMed.
- (a) V. K. Pandey and P. Anbarasan, J. Org. Chem., 2014, 79, 4154 CrossRef CAS PubMed; (b) P. Saravanan and P. Anbarasan, Adv. Synth. Catal., 2015, 357, 3521 CrossRef CAS.
- (a) M. D. Hossain, Y. Ikegami and T. Kitamura, J. Org. Chem., 2006, 71, 9903 CrossRef CAS PubMed; (b) M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 73, 4602 CrossRef CAS PubMed.
- B. Wang, J. W. Graskemper, L. Qin and S. G. DiMagno, Angew. Chem., Int. Ed., 2010, 49, 4079 CrossRef CAS PubMed.
- A. I. Konovalov, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 13410 CrossRef CAS PubMed.
- N. Nebra and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 16998 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, optimization, characterizations data, 19F NMR and GCMS spectra. See DOI: 10.1039/c5ra27128b |
| This journal is © The Royal Society of Chemistry 2016 |