
Solvent tunable photophysics of acridone: a quantum chemical perspective
Abstract
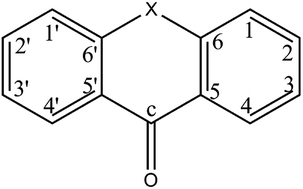
High-level electronic structure methods and quantum chemistry programs have been employed for a thorough investigation of the photophysics of acridone in isolated and solvated states. A kaleidoscope of photophysical behavior results by varying the medium in which acridone finds itself. With the computed intersystem crossing rate constants of the order of 1011 s−1 in vacuum, the photoexcited species acts as an effective triplet sensitizer. In polar, aprotic media, the radiationless decay processes for the photoexcited singlet state are still predominant, however, a delayed fluorescence may also be observed. Such delayed fluorescence comes into play due to the near degeneracy of the 1(πHπ*L) and 3(nOπ*L) electronic states in such a solvent. As the polarity, as well as the proticity of the solvent increases, the energy shifts experienced by the involved electronic states cause the photoexcited molecule to relax primarily via fluorescence emission to the ground state with a rate constant of the order of about 107 s−1. In such solvents, acridone is a good fluorescence marker. Hence, tuning the polarity and proticity of the solvent should turn acridone into a dark (triplet formation) or a bright (fluorescence) sensitizer.
 Please wait while we load your content...
Please wait while we load your content...

