DOI:
10.1039/C5RA27018A
(Paper)
RSC Adv., 2016,
6, 12414-12421
Synthesis of protein/hydroxyapatite nano-composites by a high-gravity co-precipitation method
Received
17th December 2015
, Accepted 19th January 2016
First published on 22nd January 2016
Abstract
This study explored potential use of a high-gravity co-precipitation strategy to synthesize gelatin/hydroxyapatite (Gel/HAP) nano-composites with high protein adsorption efficiencies. The physicochemical properties were characterized using X-ray diffraction, transmission electron microscopy, Fourier transform infrared spectra, thermal gravimetric analysis and Brunauer–Emmett–Teller. Biocompatibility was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assays on hFOB 1.19 osteoblast cells. The results confirmed formation of Gel/HAP composites exhibiting nano-rod-like morphologies with high protein adsorption efficiencies and biocompatibilities. In addition, the Gel/HAP nano-composites were molded into cylindrical shaped Calcium Phosphate Cements (CPC), displaying an average compressive modulus of ∼0.6 GPa. The compressive modulus was comparable with that of a human cancellous bone, suggestive of great potential of the Gel/HAP nano-composites in various biomedical applications through serving as bone substitute materials. The high-gravity co-precipitation technique was also applied to synthesize Silk/HAP nano-composites. Therefore, the high-gravity co-precipitation method provides a novel approach for synthesis of various protein/HAP materials.
Introduction
There is a great demand for synthetic bone substitute materials because of various diseases.1,2 Considering that natural bones are composites of mainly hydroxyapatite (HAP) nano-crystals embedded in collagen (Col) fibrils,3 research interest has focused on fabrication of bone-mimetic nano-composites for bone substitute applications, such as Col/HAP.4 Due to low solubility and expensive price of Col, gelatin (Gel), a denatured form of Col, has been a favoured substitution of Col. With respect to Col, Gel has many advantages such as inexpensive, commercially available, dispersible in water, easy for chemical modification, nontoxic, completely resorbable in vivo, low immunogenicity and FDA approval. Hence, Gel/HAP composites have received considerable attention and been extensively investigated as promising bone replacement biomaterials for hard tissue repairing.5–11
Many researchers have carried out various strategies to prepare Gel/HAP composites.12–16 Gel/HAP composites have been prepared by dispersing HAP powders in Gel solutions. A major disadvantage of this method was that the HAP could not disperse well in Gel and would aggregate.17,18 To overcome the disadvantage, researchers developed biomimetic processes to prepare Gel/HAP composites, by co-precipitation of HAP in Gel solutions, very similar to biological formation of natural bones.19,20 Conventionally, the co-precipitation involved gradually adding small droplets of phosphate salt solution into a bulk solution containing pre-mixed Gel and calcium salts. However, the addition of a small droplet into a bulk solution might cause a heterogeneous distribution of the phosphate groups, which would usually make an unfavourable long period of continuously and vigorously stirring necessary to ensure thorough reactions. In addition, protein adsorption on inorganic materials has always been a complex paradigm,21 which made Gel adsorption on HAP a difficult process. As a result, a large amount of Gel was washed away, leading to lower protein adsorption efficiencies than theoretical values.
To encounter these problems, we devoted to develop a new rapid method for synthesis of Gel/HAP nano-composites with high protein adsorption efficiencies. High-gravity (HG) technology has been an effective chemical engineering method to intensify mass transfer in multiphase systems since 1979.22 The general idea of the HG technology is to strongly intensify substance micro-mixing under a HG environment in a rotating packed bed (RPB). In a typical HG experiment, liquid flows are pumped into the equipment and can be accelerated at a large rotating speed by the RPB. The HG process provides a strong shear stress – a centrifugal force that would split reactant solutions into tiny liquid droplets to enable better contact of the reagents and more thorough reactions between the reagents. In this way, the mass transfer between the liquid–liquid for mixing or adsorption will be largely enhanced.22 Therefore, we hypothesized that the HG technology could give better Gel adsorption performance in co-precipitation than conventional co-precipitation in stirred tank reactors (STR).
To prove our hypothesis, in this study we explored the potential of the HG technology in co-precipitation preparation of Gel/HAP nano-composites to enhance the protein adsorption. Gel/HAP nano-composites were synthesized under conventional co-precipitation in STR and under a HG field. We characterized and compared physicochemical and biocompatible properties of the Gel/HAP nano-composites under both conditions. Further, we explored potential of the Gel/HAP nano-composites in preparing Calcium Phosphate Cements (CPC), which is one of the most common materials in bone substitute applications. Moreover, we explored possibility of applying the HG co-precipitation technique to other systems, such as silk protein/hydroxyapatite (Silk/HAP) nano-composites.
Experimental
Materials and experimental setup
A schematic of experimental setup for the high-gravity (HG) co-precipitation process was shown in Fig. 1. The rotating packed bed (RPB) have been described previously and can be found elsewhere.23 The RPB mainly consisted of a rotor, a casing, two liquid inlets and a liquid outlet. Stainless wire mesh packing was installed inside the rotor. The casing has an inner diameter of 35 mm, an outer diameter of 105 mm, and an axial length of 35 mm.
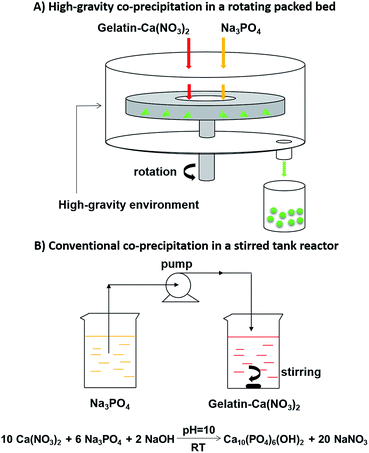 |
| | Fig. 1 Experimental setup schematics of the high-gravity (HG) co-precipitation in a rotating packed bed (A) and conventional co-precipitation in a stirred tank reactor (B) for synthesis of gelatin/hydroxyapatite (Gel/HAP) nano-composites. | |
Analytical reagent grade calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), sodium phosphate (Na3PO4), sodium hydroxide (NaOH) and absolute ethyl alcohol (CH3CH2OH) were purchased from Sinopharm Chemical Reagent Beijing Co. Ltd. Gelatin (powder from cold water fish) was purchased from Sigma-Aldrich, and used without further purification.
Raw silk (produced in Zhejiang, China) cut into small pieces were boiled in 0.05 wt% Na2CO3 for 30 min at 90 °C and washed thoroughly with deionized water for 3 times to extract natural sericin coatings. The degummed silk was dried overnight at room temperature. The dried silk was then dissolved in a Ca(NO3)2–ethanol–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:8) solution at 60 °C for 2 h. After dissolution, the silk was centrifuged at 8000 rpm for 10 min at 4 °C to remove aggregates; the supernatant was continuously dialyzed for 3 days with water changed every 5–7 h using a cellulose dialysis membrane (MWCO 8000 Da) to remove salts and any other remaining impurities from the solution. The dialyzed silk solution was then lyophilized to obtain regenerated silk protein.24
:2:8) solution at 60 °C for 2 h. After dissolution, the silk was centrifuged at 8000 rpm for 10 min at 4 °C to remove aggregates; the supernatant was continuously dialyzed for 3 days with water changed every 5–7 h using a cellulose dialysis membrane (MWCO 8000 Da) to remove salts and any other remaining impurities from the solution. The dialyzed silk solution was then lyophilized to obtain regenerated silk protein.24
Preparation of protein/hydroxyapatite
In a typical HG co-precipitation reaction, 2 L of aqueous solution containing 12.5 g of gelatin (Gel) and 0.1 M Ca(NO3)2 (Gel–Ca(NO3)2, pH = 10) was prepared. An aqueous solution of Na3PO4 (0.1 M, pH = 10) was also prepared. The two solutions were simultaneously pumped into the RPB from different inlets at a HG level of 1098 m s−2 (rotational speed at 1600 rpm (RPB1600)). The flow rates of the two solutions were set to give a Ca/P molar ratio at 1.67 under the idealized stoichiometric equation:| | |
10Ca(NO3)2 + 6Na3PO4 + 2NaOH → Ca10(PO4)6(OH)2 + 20NaNO3
| (1) |
After entering the RPB, the two solutions vigorously mixed and reacted to form Gel/HAP. The Gel/HAP was collected, aged at room temperature overnight, filtered and washed with deionized water. The product was dried at 80 °C for 12 h, then ground into powder to obtain Gel/HAP composites (Gel/HAPRPB1600). Silk/HAP was prepared the same way as Gel/HAP by replacing Gel with the regenerated silk protein.
For comparison purposes, Gel/HAP composites were also prepared by a conventional co-precipitation method in a stirred tank reactor (STR) by a modified precipitation method.25 Aqueous solutions of Gel–Ca(NO3)2 and Na3PO4 were prepared in the same way as that in the HG co-precipitation reaction. The Na3PO4 solution was added into the Gel–Ca(NO3)2 solution drop by drop (4 mL min−1) under vigorously stirring. Volumes of the two solutions were calculated to give a Ca/P molar ratio at 1.67. After completing the addition of the Na3PO4 solution, the suspension was continually stirred for another 2 h to form Gel/HAP. The Gel/HAP was aged at room temperature overnight, filtered and washed with deionized water, followed by drying at 80 °C for 12 h and then grounding into powder to obtain Gel/HAP composites (Gel/HAPSTR).
Preparation of calcium phosphate cements (CPC)
Both the Gel/HAPRPB1600 and Gel/HAPSTR were separately shaped into cylindrical samples of ∼13 mm in diameter and ∼10 mm in height using a powder tablet machine (FW-4A, TIAN JIN TUO PU INSTRUMENTS CO., LTD) under compression pressure of 5 MP for 5 min. The samples were removed from the cylindrical mold and used for mechanical tests.
Physicochemical characterizations of Gel/HAP
X-ray diffraction analysis (XRD). Crystal form of the Gel/HAP was examined by XRD analysis (XRD-6000, Shimadzu, Japan). The experiments were operated at voltage and current setting of 40 kV and 30 mA, respectively. The XRD patterns were recorded in a range of 5° to 90° at a scan rate of 5° min−1 with Cu Kα (λ = 0.15406 nm) radiation. Crystallographic identification of the XRD patterns was accomplished by comparing with standard data compiled by the International Centre for Diffraction Data (ICDD).
Transmission electron microscopy (TEM). TEM experiments were performed on an H-800 (Hitachi, Japan). The Gel/HAP nano-composites were dispersed in absolute ethyl alcohol and samples for TEM characterization were prepared by evaporating a droplet of dispersion on a Cu grid. The morphology and size of the Gel/HAP were investigated. Diameters and lengths and corresponding distributions were derived from more than 100 particles in TEM images using ImageJ (NIH, USA).
Fourier transform infrared spectra (FTIR). Chemical bonding in the Gel/HAP was analysed using FTIR. The Gel/HAP was mixed with KBr in a mass ratio of 1:100 and compressed (10 MPa) to form disks at room temperature for FTIR analysis, which was carried out on an FTIR instrument (FTIR, Bruker Optics VERTEX70, USA) in the transmission mode in a range of 400 to 4000 cm−1 with a resolution of 4 cm−1.
Thermal gravimetric analysis (TGA). Thermal stability and chemical composition of the Gel/HAP were evaluated by thermal gravimetric analysis (TGA/DSC 1/1100 SF, METTLER TOLEDO, USA). Approximately 5–10 mg of sample was ground with a mortar and pestle and then placed in a ceramics sample pan for thermal analysis. The TGA measurements were carried out under nitrogen atmosphere at a heating rate of 5 °C min−1 in a temperature range of 20 to 800 °C. Gel contents in the Gel/HAP composites (Gel adsorption efficiency) were calculated following| | |
Gelatin (%) = weight loss of Gel/HAP composites − weight loss of HAP (%)
| (2) |
Brunauer–Emmett–Teller (BET). BET surface areas of the HAP nanoparticles were determined by a surface area and porosity analyser (Quantachrome Instruments). The samples were heated at 80 °C for 4 h in a vacuum before the test in order to remove absorbed vapour and air.
Mechanical tests
Compressive tests were conducted to evaluate mechanical performance of the Gel/HAP-based CPC. The tests were carried out with an Instron-1185 mechanical testing machine with a load cell of 10 kN at a cross-head speed of 2 mm min−1 (as required by GB8813-88 ISO844-1978) until failure. Compressive moduli were calculated from slope of the initial linear portion of the stress–strain curves at 5% of strain. For experimental convenience, all tests were done at room temperature.
Cell culture and MTT-based cytotoxicity assay
Osteoblast cells (hFOB 1.19) were purchased from Xiehe Cell Resource Center (Xiehe, Beijing). The cells were cultured according to the recommended protocol in 1:1 mixture of Ham's F12 Medium Dulbecco's Modified Eagle's Medium (Xiehe), with 2.5 mM L-glutamine, supplemented with 0.3 mg mL−1 G418 (Xiehe) and 10% fetal bovine serum (Xiehe). Cells were maintained at 34 °C with 5% CO2, fed every 2–3 days and passaged according to the protocol.
To assess cytotoxicity of the Gel/HAP, hFOB 1.19 cells were seeded in a standard 96-well plate at a density of 100000 cells per well (100 μL) in triplicate. After 24 h, various amounts of the Gel/HAP were added into the wells to give final concentrations of up to 1000 μg mL−1. Cells were incubated for another 24 h, then used in the cytotoxicity assay. For the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay, 50 μL of the MTT solution (2.5 mg mL−1) was added to each experimental well, including controls (cells seeded without addition of the Gel/HAP). The plate was then returned to the cell culture incubator. After 4 h of incubation, the wells were aspirated, followed by addition of 150 μL of dimethyl sulfoxide (DMSO) to each well. The plate cover was then removed and absorbance in each well was measured at 570 nm using a microtiter plate reader. The average absorbance values from triplicate readings were determined. Cell viability was calculated following
| | |
Cell viability (%) = (ABSe − ABSb)/(ABS0 − ABSb) × 100
| (3) |
where ABS
e was the average values from experimental wells, ABS
0 was the average values from control wells and the average value for the blank (ABS
b) subtracted. Cell viability values which were lower than 100% indicated a reduction in cell proliferation rate.
Statistical analysis
All data were presented as means plus or minus standard deviation arising from measurements made from at least three independent experiments. Statistical analysis was performed on cell data using an unpaired Student's t-test with a confidence level of 0.05 unless otherwise noted.
Results and discussions
Preparation of gelatin/hydroxyapatite (Gel/HAP) nano-composites
Ca(NO3)2 and Na3PO4 were used as calcium and phosphorous sources, respectively. Gel was dissolved in the Ca(NO3)2 solution to get a pre-mixed solution of Gel and Ca(NO3)2 (Gel–Ca(NO3)2). A schematic of the experimental setup for the high-gravity (HG) co-precipitation was shown in Fig. 1. The principle component was a rotating packed bed (RPB). The two reactant solutions of Gel–Ca(NO3)2 and Na3PO4 were simultaneously pumped into the RPB from different inlets. The solutions were spread and split into tiny liquid droplets, vigorously mixed and reacted, via a centrifugal force generated by the RPB. In this way, the HG co-precipitation enabled better mixing and contact of the reagents, leading to more thorough reactions between the reagents than conventional co-precipitation in stirred tank reactors (STR).22,26,27 The Gel/HAP composites synthesized by the HG co-precipitation were labelled as Gel/HAPRPB1600. Gel/HAP composites were also prepared by a conventional co-precipitation method to serve as a control (Gel/HAPSTR).
Characterizations of the Gel/HAP nano-composites
X-ray diffraction analysis (XRD). XRD patterns of the Gel/HAP were shown in Fig. 2. Characteristic peaks of HAP were identified for the Gel/HAPRPB1600, corresponding to (002), (102), (210), (211), (112), (300), (202), (310), (222), (213), (004) lattice planes, respectively, according to standard JCPDS (09-0432).25,28 The results not only confirmed formation of HAP, but also indicated that addition of Gel did not change crystalline characteristics of the HAP.29,30 However, the predominant diffraction peaks of HAP were obscure in the XRD pattern of the Gel/HAPRPB1600, indicating low crystallinity. In addition, crystalline peaks of the Gel/HAPSTR (Fig. 2) were at the same positions as that of the Gel/HAPRPB1600, suggesting that the HG co-precipitation did not cause phase difference from the conventional co-precipitation.
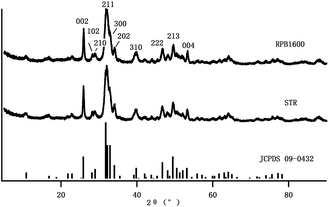 |
| | Fig. 2 XRD patterns of the Gel/HAPRPB1600 and Gel/HAPSTR. The diffraction peaks of the HAP structures were confirmed and labelled according to standard card of HAP (JCPDS 09-0432). | |
Transmission electron microscopy (TEM). Fig. 3 showed morphology of the Gel/HAP observed by TEM. As it may be seen, rod shaped HAP particles were embedded in Gel matrix. We analysed more than 100 of the Gel/HAPRPB1600 particles and found that the Gel/HAPRPB1600 were 6.5 ± 2.0 nm in diameter and 23.9 ± 8.9 nm in length. The diameters and lengths of the Gel/HAPRPB1600 distributed in the range of 3.3–18.2 and 9.4–47.7 nm, respectively. Meanwhile, the diameters of the Gel/HAPSTR were between 3.4 and 18.6 nm with an average of 7.2 ± 3.2 nm and the lengths were between 9.5 and 61.6 nm with an average of 25.1 ± 10.4 nm. These results suggested that the Gel/HAPRPB1600 exhibited similar morphology as observed in the Gel/HAPSTR synthesized by conventional co-precipitation.29 It is of note that, our previous study showed that HAP nanoparticles synthesized by HG precipitation (HAPRPB1600) were 1.9–14.2 nm wide and 4.0–36.9 nm long, while HAP nanoparticles synthesized by conventional precipitation (HAPSTR) were 4.2–20.6 nm wide and 10.9–91.6 nm long.23 Considering the smaller size of the HAPRPB1600 than that of the HAPSTR and the similar size of the Gel/HAPRPB1600 to that of the Gel/HAPSTR, it could be deduced that the Gel/HAPRPB1600 possessed thicker Gel coatings than the Gel/HAPSTR. In other words, this result implied enhanced Gel incorporation by the HG co-precipitation over the conventional co-precipitation.
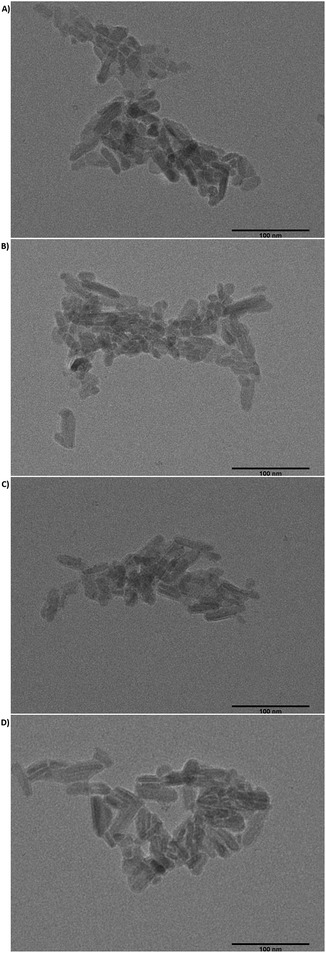 |
| | Fig. 3 TEM images of the Gel/HAPRPB1600 (A and B) and Gel/HAPSTR (C and D) at magnification ×50000. The scale bars were 100 nm. For the Gel/HAPRPB1600, diameters ranged from 3.3–18.2 nm with an average value of 6.5 ± 2.0 nm, and lengths ranged from 9.4–47.4 nm with an average value of 23.9 ± 8.9 nm (n = 128). Meanwhile, for the Gel/HAPSTR, diameters ranged from 3.4–18.6 nm with an average value of 7.2 ± 3.2 nm, lengths ranged from 9.5–61.6 nm with an average value of 25.1 ± 10.4 nm (n = 128). | |
Fourier transform infrared spectra (FTIR). The FTIR spectra of the Gel/HAPRPB1600 and Gel/HAPSTR were shown in Fig. 4. FTIR band 3600–3200 cm−1 due to H2O adsorption was observed. Characteristic bands of Gel were observed. The band at 3086 cm−1 was ascribed to N–H stretching, 2960 and 2883 cm−1 to C–H stretching vibration, 1668 cm−1 to N–H stretching (amide I), 1545 cm−1 to N–H stretching, N–H in-plane bending and C–H stretching (amide II) and 1230 cm−1 to N–H, C–N and N–H in-plane stretching (amide III). The bands, the same as those observed in pure Gel previously reported,10,30,31 confirmed presence of Gel in the Gel/HAPRPB1600. With regard to HAP, characteristic bands at 1097 cm−1 (asymmetrical stretching ν3), 1037 cm−1 (asymmetrical stretching ν3), 958 cm−1 (symmetric stretching ν1), 604 cm−1 (bending ν4), 565 cm−1 (bending ν4) and 474 cm−1 (symmetric stretching ν2) were attributed to vibrational modes of PO43− in the HAP phase. The bands confirmed existence of HAP in the Gel/HAPRPB1600. Especially, the band at 1037 cm−1 indicated a good stoichiometric composition of the HAP phase.31 In addition, bands at approximately 3573 cm−1 and 633 cm−1 were observed in the spectrum. They belonged to stretching vibration and bending deformation of structural OH– group in HAP phase, demonstrating that the inorganic phase in the Gel/HAPRPB1600 was HAP. CO32− derived band was also observed at 872 cm−1, indicating incorporation of CO32− in the HAP phase. The band was consistent with previous studies and the carbonate was mostly caused by adsorption of CO2 from atmosphere.29 More importantly, bands related to interaction between Gel and HAP were observed in the range of 1300–1450 cm−1. These bands indicated formation of chemical bonds between a carboxyl group COO− of Gel and Ca2+ of HAP (Ca–COO−).32 The FTIR spectra of Gel/HAPSTR showed characteristic bands of Gel and HAP, similar to that of Gel/HAPRPB1600. However, the intensity of the bands was weaker than that of Gel/HAPRPB1600. It was reported that the amount of Gel influenced the FTIR band intensity with increasing intensity as the Gel concentration increased.10 Therefore, more pronounced bands in Gel/HAPRPB1600 also suggested enhanced Gel incorporation by the HG co-precipitation over the conventional co-precipitation.
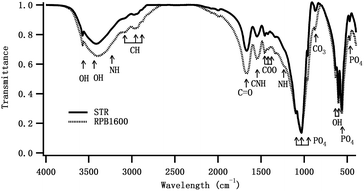 |
| | Fig. 4 FTIR spectra for Gel/HAPRPB1600 and Gel/HAPSTR. Peaks at 3573, 3600–3200, 3086, 2960, 2883, 1668, 1545, 1300–1450, 1097, 1037, 958, 872, 633, 604, 565, 474 cm−1 were labelled with arrows from left to right and described with more details in the text. | |
Thermal gravimetric analysis (TGA). To characterize thermal stability and chemical composition of the Gel/HAP, TGA was performed. As shown in Fig. 5, weight loss was observed at three temperature ranges, about 100–120, 250–420 and 450–700 °C. Weight loss between 100 and 120 °C corresponded to evaporation of volatile components such as H2O from the Gel/HAPRPB1600. Degradation of Gel began at ∼250 °C, while degradation of pure Gel was previously reported to start from 193 °C. This result strongly indicated existence of chemical bonds between HAP and Gel in the Gel/HAPRPB1600.25 In addition, it was reported that Gel usually achieved fully decomposition below 420 °C,25 while weight loss was also observed between 450 and 700 °C in the Gel/HAPRPB1600. This result also indicated existence of Gel that combined with HAP in the Gel/HAPRPB1600. Besides, TGA showed weight loss as a function of temperature from which the content of adsorbed Gel could be calculated. Total weight loss of the Gel/HAPRPB1600 (18.0 ± 3.2%) was less than the theoretical content of Gel (38%), implying that only part of the Gel in the starting solution was incorporated into the composites.25 The Gel/HAPSTR was also analysed by TGA. However, weight loss of the Gel/HAPSTR (13.6 ± 3.9%) was less than that in the Gel/HAPRPB1600. Therefore, it was reasoned that the Gel adsorbed into the Gel/HAPRPB1600 was more than that into the Gel/HAPSTR. This result was consistent with the FTIR results. In this sense, the HG co-precipitation enhanced Gel adsorption in synthesizing Gel/HAP nano-composites than the conventional co-precipitation.
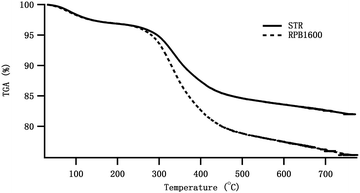 |
| | Fig. 5 TGA measurements of the Gel/HAPRPB1600 and Gel/HAPSTR in the temperature range of [20, 800] °C. Weight loss was observed at temperature ranges of 100–120, 250–420 and 450–700 °C. | |
Biocompatibility of the Gel/HAPRPB1600. From the results discussed above, it could be concluded that the Gel adsorption on the HAP could be enhanced by the HG technology. Although the HG technology was proven to be efficient in rapid production of Gel/HAP nano-composites, the question of whether or not the properties of the as-prepared nano-composites under the HG field is affected compared to the conventional co-precipitation method remains to be clarified. Therefore, we assessed cytotoxicity of the Gel/HAPRPB1600 by determining cell viability in the presence of up to 1000 μg mL−1 of the Gel/HAPRPB1600 (Fig. 6). Osteoblast cells (hFOB 1.19) were utilized throughout cell toxicity assays. The result clearly showed excellent viability of cells (>80%), indicating good cytocompatibility of the Gel/HAPRPB1600 within the range of concentration tested. The cell viability in the presence of the Gel/HAPRPB1600 was better than that of Gel/HAPSTR (>70%), especially at higher nano-composite concentrations, indicating enhanced cytocompatibility under a HG field over the conventional co-precipitation. Previous studies have reported that particle sizes, morphologies and surface properties affected cellular responses.33–38 In the present study, the enhanced cytocompatibility could be explained from several aspects, including different Gel adsorption efficiencies, particle sizes and surface areas. Firstly, FTIR and TGA analysis demonstrated a higher Gel adsorption efficiency of the Gel/HAPRPB1600 than that of Gel/HAPSTR. Secondly, TEM results showed smaller particle sizes of the Gel/HAPRPB1600 than that of Gel/HAPSTR. Thirdly, the Gel/HAPRPB1600 displayed a large surface area (41.7 ± 4.8 m2 g−1) than that of Gel/HAPSTR (31.2 ± 5.6 m2 g−1). All of the above factors would affect activity of the adsorbed Gel on the HAP surfaces, and thus influence the interaction between the Gel/HAP with the cells.
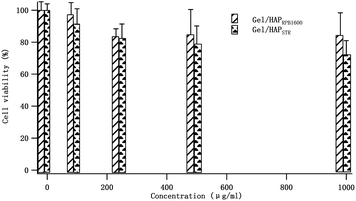 |
| | Fig. 6 Cell viabilities of hFOB 1.19 osteoblast cells in the presence of the Gel/HAPRPB1600 and Gel/HAPSTR. A comparable number of cells for concentrations at 0, 100, 250, 500 and 1000 μg mL−1 was obtained after 24 h incubation. Data were normalized to that without added reagents (0 μg mL−1). | |
Use of the Gel/HAPRPB1600 in preparing calcium phosphate cements (CPC)
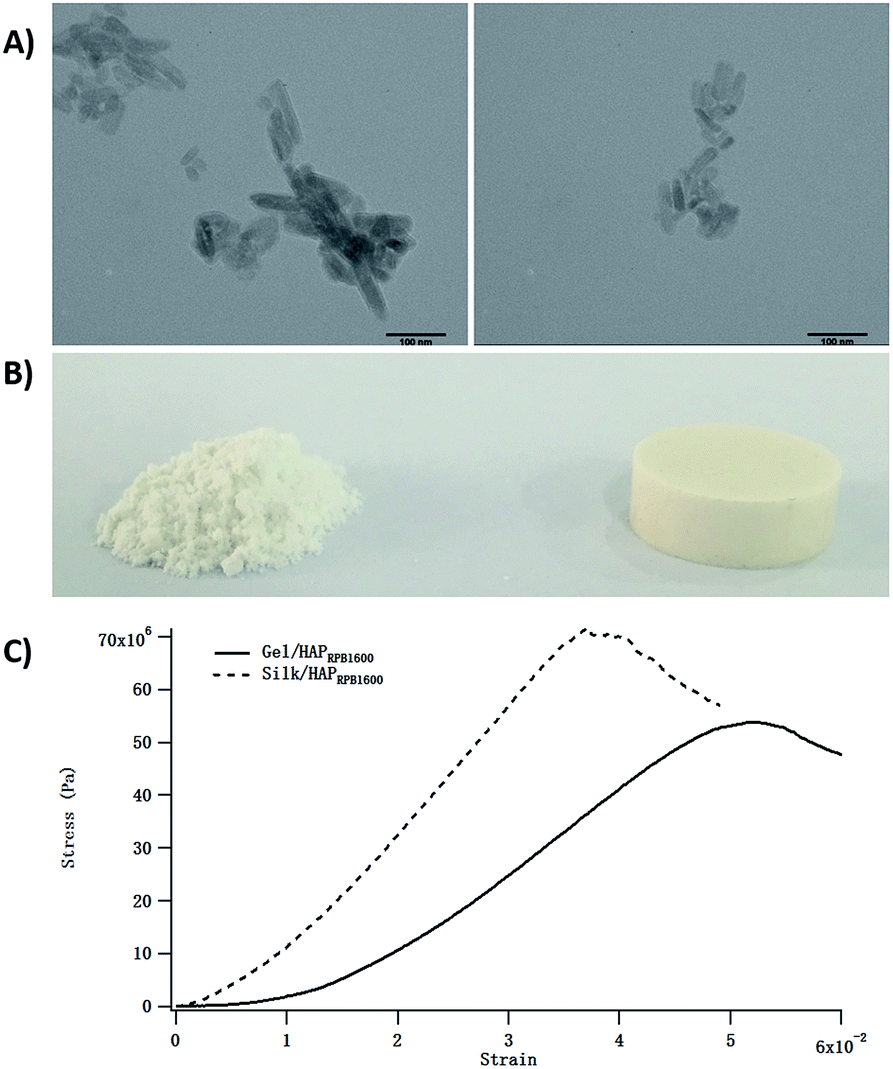
The excellent cytocompatibility implied great potential of using the Gel/HAPRPB1600 in biomedical applications. To explore possibility of applying the Gel/HAPRPB1600 as bone substitute materials, we used the Gel/HAPRPB1600 in preparing CPC, which were one of the most common bone substitute materials.39,40 The Gel/HAPRPB1600 were molded into cylindrical samples and characterized in mechanical tests (Fig. 7). Compressive modulus of the Gel/HAPRPB1600-based CPC was measured to be 0.6 ± 0.1 GPa (n = 4) at 5% strain. The compressive modulus of the Gel/HAPRPB1600-based CPC was comparable to that of natural trabecular bones,31 which demonstrated great potential of the Gel/HAPRPB1600 in fabricating new materials such as bone fillers.41 Gel/HAPSTR-based CPC were also prepared, displaying an average compressive modulus of 1.1 ± 0.1 GPa (n = 4). The compressive modulus of the Gel/HAPSTR-based CPC was higher than that of the Gel/HAPRPB1600-based CPC, which might be explained by the lower Gel content in the Gel/HAPSTR as indicated by the TEM, FTIR and TGA results. It has been reported that compressive modulus of HAP would be significantly enhanced upon addition of Gel. Gel could serve as “glue” that bonds HAP. But the enhancement effect did not increasing linearly with increasing the amount of Gel. Although presence of up to 5% of Gel could improve mechanical properties of Gel/HAP composites, Gel contents greater than 5% would decrease the compressive strength.30
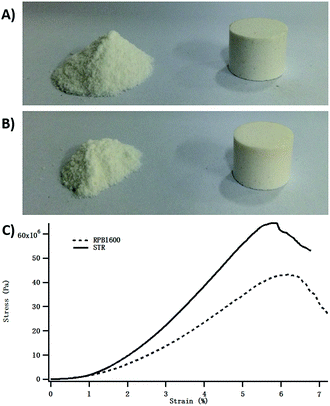 |
| | Fig. 7 Calcium phosphate cements (CPC) prepared from the Gel/HAPRPB1600 (A) and Gel/HAPSTR (B) and typical stress–strain curves in mechanical tests (C). Compressive moduli at 5% strain of the Gel/HAPRPB1600-based and Gel/HAPSTR-based CPC were 0.6 ± 0.1 GPa (n = 4) and 1.1 ± 0.1 GPa (n = 4), respectively. | |
To prove our explanation, we investigated the effect of Gel contents on the mechanical properties of Gel/HAPRPB1600-based CPC. More Gel/HAPRPB1600-based CPC were prepared by tuning the initial Gel contents (0%, 10%, 20%, 30% and 38%). As shown in Fig. 8, the compressive moduli of Gel/HAPRPB1600-based CPC with different initial Gel content were all larger than that of CPC without Gel (0%). As the initial Gel content increased from 0% to 10%, the compressive modulus increased. However, as the initial Gel content further increased from 10% to 38%, the compressive modulus decreased. The initial Gel content of 10% was found to be the optimal for preparing Gel/HAPRPB1600-based CPC. Because the initial Gel content could not represent the precise Gel content, we calculated Gel adsorption efficiencies from TGA analysis results. As Fig. 9 showed, the optimal initial Gel content of 10% corresponded to an efficiency of ∼5%, which was consistent with previous report.30 Beside of proving our explanation, it is also interesting to notice that the (initial content)–(adsorption efficiency) relation could be described reasonably well by a linear fit (Fig. 9), suggesting an intrinsic relation between the initial Gel content and Gel adsorption efficiency under HG fields.
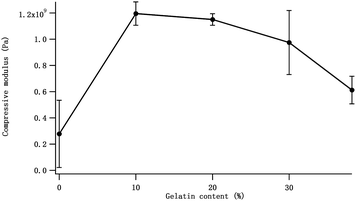 |
| | Fig. 8 The effect of the initial Gel content on the compressive modulus of the Gel/HAPRPB1600-based CPC. | |
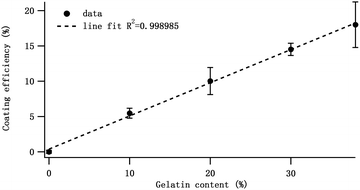 |
| | Fig. 9 The Gel adsorption efficiency of the Gel/HAPRPB1600 prepared at different initial Gel content. The function could be fitted to a linear equation with R2 = 0.998985. | |
Utilization of the HG co-precipitation in synthesizing silk protein/hydroxyapatite (Silk/HAP) nano-composites
After we explored the potential of the HG co-precipitation in synthesizing Gel/HAP nano-composites, we went a step further to explore whether this technology could be applied to other systems. Among various proteins used in HAP-based composites, silk protein is one of the most extensively studied. Herein, we used a model system-Silk/HAP. Silk/HAP was prepared the same way as Gel/HAPRPB1600 by replacing Gel with the regenerated silk protein. TEM results clearly demonstrated successful production of Silk/HAP nano-composites (Fig. 10A). The Silk/HAP displayed diameters and lengths between 4.7–27.0 and 19.0–105.8 nm, respectively. The Silk/HAP nano-composites were also molded into cylindrical CPC (Fig. 10B). The compressive modulus at 5% strain calculated from stress–strain curves (Fig. 10C) was found to be 1.2 ± 0.2 GPa.
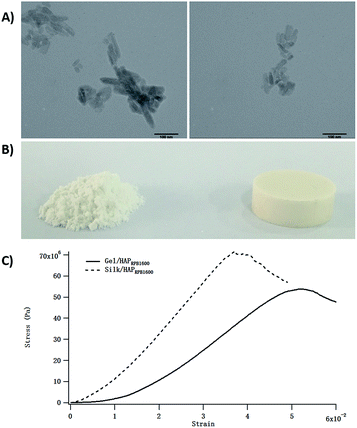 |
| | Fig. 10 TEM images of the Silk/HAP nano-composites (A) displaying diameters ranged from 4.7–27.0 nm with an average value of ∼13.0 nm, and lengths ranged from 19.0–105.8 nm with an average value of ∼43.7 nm, respectively. The scale bars were 100 nm. CPC prepared from Silk/HAP nano-composites (B) and typical stress–strain curves in compression tests (C). | |
Conclusions
This study explored the potential use of a high-gravity (HG) co-precipitation method to prepare gelatin/hydroxyapatite (Gel/HAP) nano-composites with high protein adsorption efficiencies. Nano-rod shaped Gel/HAP composites with a diameter distribution of 3.3–18.2 nm and a length distribution of 9.4–47.7 nm were successfully obtained. A high Gel adsorption efficiency over that in the conventional co-precipitation was observed. Enhanced biocompatibilities with hFOB 1.19 osteoblast cells were also observed. The compressive modulus of Calcium Phosphate Cements (CPC) based on the resultant Gel/HAP nano-composites was ∼0.6 GPa, comparable to that of trabecular bones. These findings demonstrated great potential of the newly synthesized Gel/HAP nano-composites to be used as bone substitute materials for bone repairing. The high-gravity co-precipitation method provides a new approach for synthesis of Gel/HAP nano-composites and opens new possibilities for producing various other protein–inorganic nano-composite systems which require higher protein adsorption.
Acknowledgements
The author would like to thank Dr Yuan Le and Mr Mengtao Liu for experimental support on the cell assay, thank Dr Zhiping Liu for help on the cement forming molds. The work is supported by the Foundation of Beijing University of Chemical Technology (12060029009, 12060046001), the Foundation of State Key Laboratory of Organic-inorganic Composites (201311, 201403002), the Fundamental Research Funds for the Central Universities (YS1401) and the National Natural Science Foundation of China (Grant no. 31400813).
Notes and references
- J. D. Currey, Bones: structure and mechanics, Princeton University Press, Princeton [N.J.], 2nd edn, 2002 Search PubMed.
- S. Mann, Biomineralization: principles and concepts in bioinorganic materials chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- D. G. T. Strange and M. L. Oyen, Acta Biomater., 2011, 7, 3586–3594 CrossRef CAS PubMed.
- A. Tsetsekou, D. Brasinika, V. Vaou and E. Chatzitheodoridis, Mater. Sci. Eng., C, 2014, 43, 555–565 CrossRef CAS PubMed.
- A. Zaupa, A. T. Neffe, B. F. Pierce, A. Lendlein and D. Hofmann, Int. J. Artif. Organs, 2011, 34, 139–151 CrossRef CAS PubMed.
- A. Vyalikh, P. Simon, T. Kollmann, R. Kniep and U. Scheler, J. Phys. Chem. C, 2011, 115, 1513–1519 CAS.
- A. T. Neffe, A. Zaupa, B. F. Pierce, D. Hofmann and A. Lendlein, Macromol. Rapid Commun., 2010, 31, 1534–1539 CrossRef CAS.
- M. Dressler, E. Dombrowski, U. Simon, J. Bornstein, V. D. Hodoroaba, M. Feigl, S. Grunow, R. Gildenhaar and M. Neumann, J. Eur. Ceram. Soc., 2011, 31, 523–529 CrossRef CAS.
- G. Tronci, A. T. Neffe, B. F. Pierce and A. Lendlein, J. Mater. Chem., 2010, 20, 8875–8884 RSC.
- R. K. Brundavanam, Z.-T. Jiang, P. Chapman, X.-T. Le, N. Mondinos, D. Fawcett and G. E. J. g. p. m. e. a. Poinern, Ultrason. Sonochem., 2011, 18, 697–703 CrossRef CAS PubMed.
- K. Miura, T. Anada, Y. Honda, Y. Shiwaku, T. Kawai, S. Echigo, T. Takahashi and O. Suzuki, Appl. Surf. Sci., 2013, 282, 138–145 CrossRef CAS.
- K. J. Roche and K. T. Stanton, J. Cryst. Growth, 2015, 409, 80–88 CrossRef CAS.
- S. Teng and P. Wang, Mater. Lett., 2011, 65, 1348–1350 CrossRef CAS.
- J. H. An, O. S. Han, D. H. Kohn, Y. J. Park and H. J. Song, J. Nanosci. Nanotechnol., 2015, 15, 5668–5671 CrossRef CAS PubMed.
- S. N. Danilchenko, O. V. Kalinkevich, M. V. Pogorelov, A. N. Kalinkevich, A. M. Sklyar, T. G. Kalinichenko, V. Y. Ilyashenko, V. V. Starikov, V. I. Bumeyster, V. Z. Sikora and L. F. Sukhodub, J. Biomed. Mater. Res., Part A, 2011, 96, 639–647 CrossRef PubMed.
- Z. Su, J. Li, Z. Ouyang, M. M. L. Arras, G. Wei and K. D. Jandt, RSC Adv., 2014, 4, 14833–14839 RSC.
- M. Zandi, H. Mirzadeh, C. Mayer, H. Urch, M. B. Eslaminejad, F. Bagheri and H. Mivehchi, J. Biomed. Mater. Res., Part A, 2010, 92, 1244–1255 Search PubMed.
- M. Azami, M. Rabiee and F. Moztarzadeh, Polym. Compos., 2010, 31, 2112–2120 CrossRef CAS.
- B. Basu, S. K. Swain and D. Sarkar, RSC Adv., 2013, 3, 14622–14633 RSC.
- J. Lee, B. Choi, J. Choi, G. Kim, D. Hwang, M. Chang, J. Byun and U. Kim, Tissue Eng. Regener. Med., 2013, 10, 47–52 CrossRef CAS.
- V. O. Kollath, S. Mullens, J. Luyten, K. Traina and R. Cloots, RSC Adv., 2015, 5, 55625–55632 RSC.
- J. Chen, Y. Wang, F. Guo, X. Wang and C. Zheng, Ind. Eng. Chem. Res., 2000, 39, 948–954 CrossRef CAS.
- H. Peng, J. Wang, S. Lv, J. Wen and J. Chen, Ceram. Int., 2015, 41, 14340–14349 CrossRef CAS.
- H. Wang and Y. Zhang, Sci. Rep., 2014, 4 Search PubMed.
- W. Wei, R. Sun, Z. Jin, J. Cui and Z. Wei, Appl. Surf. Sci., 2014, 292, 1020–1029 CrossRef CAS.
- A. J. Nathanaela, S. I. Hong, D. Mangalaraj and P. C. Chen, Chem. Eng. J., 2011, 173, 846–854 CrossRef.
- Q. Yang, J. Wang, F. Guo and J. Chen, Ind. Eng. Chem. Res., 2010, 49, 9857–9863 CrossRef CAS.
- J. Chen and Y. Chang, Colloids Surf., B, 2011, 86, 169–175 CrossRef CAS PubMed.
- H. Wang, M. Bongio, K. Farbod, A. W. Nijhuis, J. van den Beucken, O. C. Boerman, J. C. van Hest, Y. Li, J. A. Jansen and S. C. Leeuwenburgh, Acta Biomater., 2014, 10, 508–519 CrossRef CAS PubMed.
- J. Chen and F. Chang, Appl. Surf. Sci., 2012, 262, 176–183 CrossRef CAS.
- S. K. Swain and D. Sarkar, Mater. Lett., 2013, 92, 252–254 CrossRef CAS.
- M. Azami, M. J. Moosavifar, N. Baheiraei, F. Moztarzadeh and J. Ai, J. Biomed. Mater. Res., Part A, 2012, 100, 1347–1355 CrossRef PubMed.
- Y. Han, S. Li, X. Cao, L. Yuan, Y. Wang, Y. Yin, T. Qiu, H. Dai and X. Wang, Sci. Rep., 2014, 4, 7 Search PubMed.
- M. Yin, Y. Yin, Y. Han, H. Dai and S. Li, J. Nanomater., 2014, 2014 Search PubMed.
- S. M. Zakaria, S. H. Sharif Zein, M. Othman and J. A. Jansen, J. Biomed. Mater. Res., Part A, 2013, 101, 1977–1985 CrossRef PubMed.
- J. Lee and H. Yun, J. Mater. Chem. B, 2014, 2, 1255–1263 RSC.
- A. J. Nathanael, R. Yuvakkumar, S. I. Hong and T. H. Oh, ACS Appl. Mater. Interfaces, 2014, 6, 9850–9857 CAS.
- C. Liao and J. Zhou, J. Phys. Chem. B, 2014, 118, 5843–5852 CrossRef CAS PubMed.
- M. Li, X. Liu, X. Liu and B. Ge, Clin. Orthop. Relat. Res., 2010, 468, 1978–1985 CrossRef PubMed.
- D. A. Oortgiesen, X. F. Walboomers, A. L. Bronckers, G. J. Meijer and J. A. Jansen, J. Tissue Eng. Regener. Med., 2014, 8, 202–209 CrossRef CAS PubMed.
- M. Peroglio, L. Gremillard, D. Eglin, P. Lezuo, M. Alini and J. Chevalier, Acta Biomater., 2010, 6, 3808–3812 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.