
Butadiene dyes based on 3-(dicyanomethylidene)indan-1-one and 1,3-bis(dicyanomethylidene)indane: synthesis, characterization and solvatochromic behaviour†
Abstract
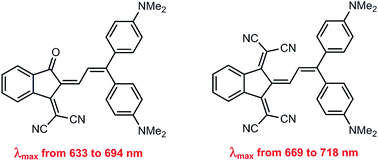
Novel donor–acceptor butadiene chromophores were synthesized by Knoevenagel condensation of different diaryl-substituted enals with 3-(dicyanomethylidene)indan-1-one and 1,3-bis(dicyanomethylidene)indane. The structures of two of these compounds were unambiguously confirmed by means of single-crystal X-ray diffraction. The absorption spectra of these dyes, as well as their solvatochromic behaviour, were studied in detail. In addition, DFT and TD-DFT quantum chemical calculations were performed to assess information regarding the topologies and absolute energies of their frontier molecular orbitals, as well as their absorption spectra.
 Please wait while we load your content...
Please wait while we load your content...

