
˙OH-initiated heterogeneous oxidation of methyl orange using an Fe–Ce/MCM-41 catalyst†
Shuaijun Wang,
Chaocheng Zhao*,
Dejun Wang,
Yongqiang Wang and
Fang Liu
College of Chemical Engineering, China University of Petroleum, Qingdao 266580, P. R. China. E-mail: zhaochch@upc.edu.cn; Tel: +86 13506360770
First published on 27th January 2016
Abstract
In this study, a simple and active Fe3O4–Fe2O3–CeO2/MCM-41 (Fe–Ce/MCM-41) catalyst was fabricated, using iron and cerium species that were simultaneously immobilized on a MCM-41 zeolite surface, and employed as a heterogeneous Fenton-like catalyst for the oxidation of methyl orange (MO) for the first time. The activity of the Fe–Ce/MCM-41 catalyst was evaluated based on the degradation of MO, and the effects of the operating conditions (i.e., pH value, catalyst addition, hydrogen peroxide (H2O2) concentration and MO concentration) on degradation performance were investigated. The kinetics results indicated that MO removal followed pseudo-first order kinetics. Based on the analysis of metal leaching, X-ray photoelectron spectroscopy, and the effects of radical scavengers, MO in the bulk solution was primarily oxidized by surface-bound ˙OH, which was generated by the reaction of Fe2+ and Ce3+ species with H2O2 on the catalyst surface. The as-prepared Fe–Ce/MCM-41 catalyst exhibited excellent performance without the aid of ultrasonic and UV light irradiation, which can decrease the operating costs. In addition, this process may be promising for application in practical wastewater treatment.
1. Introduction
In recent years, various organic wastewater pollutants have been discharged into our environment, which has endangered human health as well as the environment.1–4 Among these organic wastewater contaminants, azo dyes are of much concern due to their toxicity and refractory biological degradation.5–7 Therefore, the recovery of clean water from wastewater is increasing important.8,9Various advanced oxidation technologies, such as Fenton, ultrasonic, electrical–chemical, and photocatalytic technologies, have been proposed to treat azo dyes in wastewater.10–14 Among these wastewater remediation technologies, homogeneous Fenton oxidation, which is based on the generation of a non-selective hydroxyl radical (OH˙) for the degradation of organic pollutants, has been a popular research topic due to its high efficiency, mild reaction conditions, simple operation and environmental protection.15–19 However, a narrow pH range and the formation of iron sludge limits the practical application of homogeneous Fenton oxidation technology.20–22
To overcome the drawbacks of homogeneous Fenton oxidation, heterogeneous Fenton catalysts have been developed.12,23 In comparison to homogeneous Fenton oxidation, heterogeneous Fenton catalysts can degrade organic pollutants over a broad range of pH values with the generation of little iron sludge.24 Spinel ferrite composites and zero-valent iron have attracted considerable attention in heterogeneous advanced oxidation processes (AOPs) due to separation and recycling factors.25–27 However, most of these materials often require the aid of ultrasound and UV/visible light irradiation to achieve excellent catalytic activity, which increases the energy requirements and cost of wastewater treatment.28,29 Recently, the dispersion of metal oxides on the surface of carriers with a large surface area has been a feasible approach because it can significantly improve the catalytic activity. MCM-41 is a promising candidate for use as a carrier due to its high surface area, nontoxicity, and narrow pore size as well as its excellent physical and chemical stability.30,31
Cerium has attracted much attention due to the Ce3+/Ce4+ redox process and high oxygen storage capacity, which enhance the catalytic ability of a material for use in automotive exhaust treatment and wastewater treatment.32 Xu33 reported that nanoscaled CeO2 can enhance the performance of Fe3O4 for the oxidation of 4-chlorophenol in the presence of H2O2, and the role of CeO2 and Fe3O4 has been previously elucidated. Nevertheless, additional insight into CeO2 promotion of the activity of Fe2O3 and Fe3O4 would be beneficial. To the best of our knowledge, no previous studies have investigated the interaction between CeO2, Fe3O4, and Fe2O3 in the presence of H2O2.
Therefore, in the current study, iron oxides and cerium oxides were dispersed onto the MCM-41 surface to fabricate a new type of Fe–Ce/MCM-41 catalyst and promote the Fenton oxidation of MO by H2O2. Various characterizations have been performed, and the effects of the pH value, catalyst addition, H2O2 concentration, and temperature were investigated. In addition, the role of iron and cerium in the oxidation of MO was explored. Furthermore, a possible mechanism was proposed based on the detected ˙OH, X-ray photoelectron spectroscopy results, and metal leaching. Finally, the mineralization ability and reusability of the catalyst were evaluated.
2. Materials and methods
2.1 Preparation of catalyst
In a typical process, 1.3 g of Fe(NO3)3·9H2O and 0.45 g of Ce(NO3)3·6H2O (mole ratio of Fe(NO3)3·9H2O and Ce(NO3)3·6H2O was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dissolved in 4.3 mL of double distilled water, and then, 1.0 g of MCM-41 was added to this homogenous solution under ultrasonic oscillation (KQ-400KDB, Kunshan Ultrasonic Oscillation Instrument, Kunshan, China) with an ultrasonic frequency of 40 kHz and an ultrasonic power of 100 W for 20 min. Then, the mixture was dried at 90 °C in an oven for 4 h. Finally, Fe–Ce/MCM-41 composites with ordered mesoporous structures were obtained after calcination at 250 °C in a furnace. The iron oxide content of the as-prepared Fe–Ce/MCM-41 catalyst was 7.5 wt%.
:1) were dissolved in 4.3 mL of double distilled water, and then, 1.0 g of MCM-41 was added to this homogenous solution under ultrasonic oscillation (KQ-400KDB, Kunshan Ultrasonic Oscillation Instrument, Kunshan, China) with an ultrasonic frequency of 40 kHz and an ultrasonic power of 100 W for 20 min. Then, the mixture was dried at 90 °C in an oven for 4 h. Finally, Fe–Ce/MCM-41 composites with ordered mesoporous structures were obtained after calcination at 250 °C in a furnace. The iron oxide content of the as-prepared Fe–Ce/MCM-41 catalyst was 7.5 wt%.
2.2 Characterization
The powder X-ray diffraction (XRD) patterns of the samples were obtained using a diffractometer (X’Pert Pro, Holland) with Cu Kα radiation (40 kV, 40 mA).A Fourier transform infrared (FT-IR) spectrometer (Nicolet Nexus 670, America) was used to record the spectra of the samples in a range from 400 nm to 2500 nm.
Nitrogen adsorption/desorption was analysed using a Micromeritics ASAP 2010 sorption analyser.
The functional groups and the related oxidation state of the surface of the materials were analysed by X-ray photoelectron spectroscopy (XPS) on a PHI 5000 Versaprobe spectrometer equipped with a rotating Al anode generating Al Kα X-ray radiation at 1486.6 eV. The X-ray beam was monochromatized using seven crystals that were mounted on three Rowland circles.
The microscopic features of the samples were observed using a transmission electron microscope (TEM) (JEM 2010, JEOL, Japan) operated at 200 kV.
2.3 Degradation experiment
Batch degradation experiments with MO were carried out to explore the effects of the initial pH value, H2O2 dosage, Fe–Ce/MCM-41 amount, and MO concentration in an open 150 mL cylindrical glass tube that was maintained at a temperature of 30 ± 1 °C in a circulating water jacket. To ensure sufficient contact between the catalyst and the MO solution, a mechanical stirrer (D2004W, Shanghai SileInstrument Co., Ltd) was employed at 400 rpm. The suspended solutions were adjusted to the desired pH using 0.1 M H2SO4 or NaOH, and a specific amount of H2O2 was added to the solution to initiate the reaction. Then, the samples were collected at specific intervals and quenched with excess n-butanol. In addition, a simple magnet was employed to separate the catalyst and the MO solution. The TOC was determined using a TOC analyser (TOC-5000A, Shimadzu). After the reaction, the catalyst was collected, washed, dried, and reused with a fresh solution of MO several times.2.4 Analytical methods
The residual concentrations of the MO solution were measured with a UV-762 ultraviolet-visible spectrophotometer at 465 nm (UV-Vis 762, Shanghai Analysis Co., Shanghai). C0 represents the initial concentration, and C represents the instantaneous concentration. The concentration of ferrous ion was colorimetrically measured with 1,10-phenanthroline at 510 nm according to a previously described protocol.34 The total dissolved iron was also analysed using this method and hydroxylamine hydrochloride was used as a reducing agent.35 The dissolved cerium concentration was detected using atomic absorption spectrometry (AAS6 VARIO).3. Results and discussion
3.1 Characterization
The structures of the MCM-41 and Fe–Ce/MCM-41 catalyst were first determined using wide angle X-ray diffraction (WXRD) and the WXRD patterns are shown in Fig. 1a. As shown in Fig. 1a, the three peaks observed at 2θ values of 28.6°, 33.1° and 47.5° were assigned to the (111), (200) and (220) crystal planes, respectively, corresponding to CeO2 (ICSD collection code 61595). The peaks at 35.5°, 57.0° and 62.6° were indexed as (311), (511) and (440), respectively, which are consistent with Fe3O4 (ICSD collection code 085177). Additionally, the peaks at 24.2°, 33.1°, 35.6°, 40.9°, 49.5°, 54.1°, 62.4° and 64.0° were due to (012), (104), (110), (113), (024), (116), (214) and (300), respectively, which correspond to Fe2O3 (ICSD collection code 071194). Therefore, cerium oxides and iron oxides were successfully loaded onto the MCM-41 zeolite surface. To further investigate the structural variation of the obtained products during the fabrication process, small angle X-ray diffraction (SXRD) was performed, and the results are shown in Fig. 1b. Three peaks were observed at 2θ values of 2.30°, 3.97° and 4.60°, and can be indexed to the (100), (110) and (200) reflections, respectively, of MCM-41.30 Although the intensity of the three peaks for the Fe–Ce/MCM-41 catalyst decreased compared to those of MCM-41, the SXRD pattern of MCM-41 before and after dispersion are essentially the same. Therefore, the obtained Fe–Ce/MCM-41 catalyst has an ordered mesoporous structure and simultaneously exhibits the properties of both iron oxides and cerium oxides.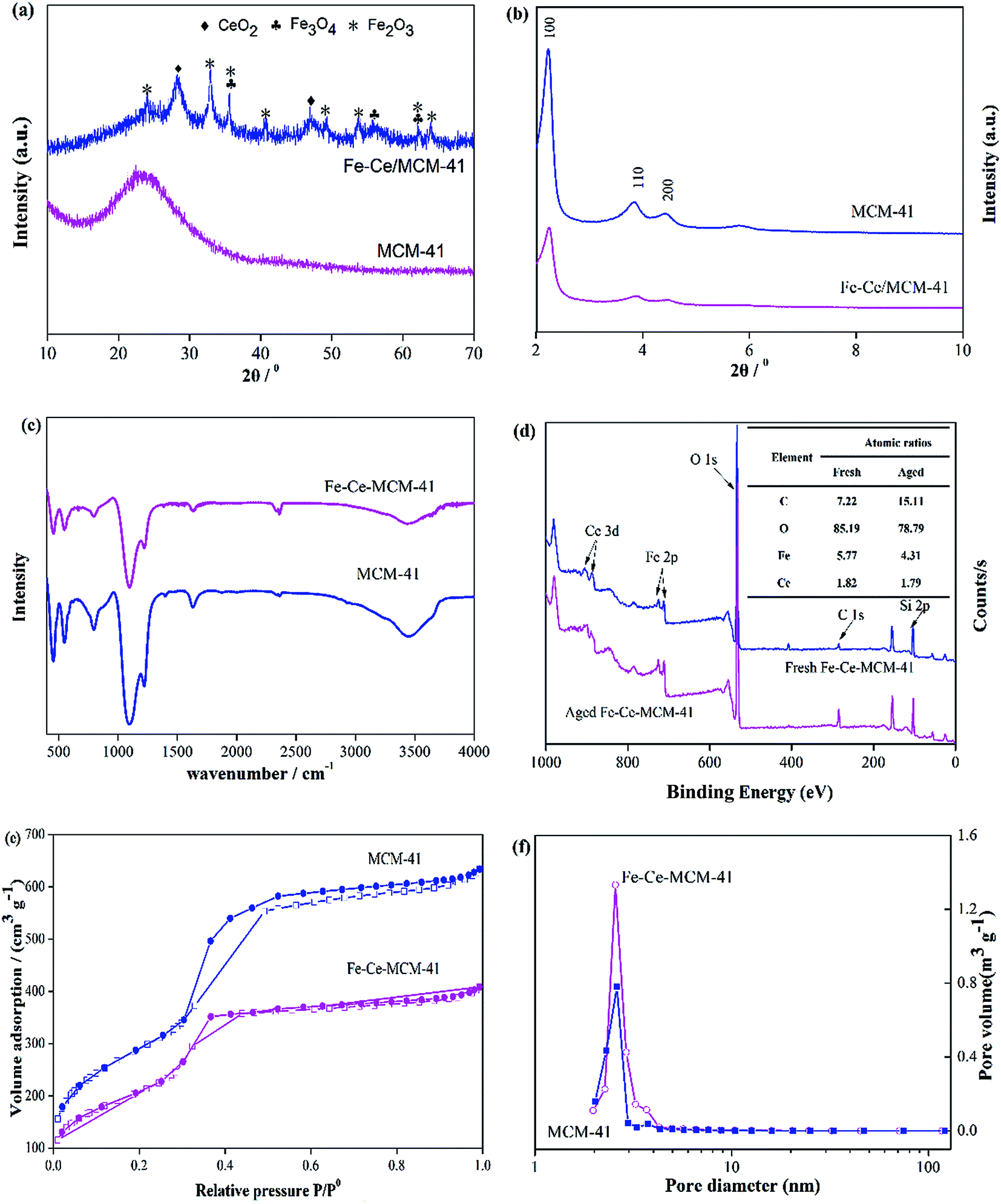 | ||
| Fig. 1 (a) WXRD patterns of ordered mesoporous MCM-41 and Fe–Ce/MCM-41. (b) SXRD patterns of ordered mesoporous MCM-41 and Fe–Ce/MCM-41. (c) FT-IR spectra of ordered mesoporous MCM-41 and Fe–Ce/MCM-41. (d) XPS analysis of the Fe–Ce/MCM-41 catalyst before and after the reaction. (e) Nitrogen adsorption/desorption isotherms of ordered mesoporous MCM-41 and Fe–Ce/MCM-41. (f) Desorption pore size distribution of ordered mesoporous MCM-41 and Fe–Ce/MCM-41. | ||
The FT-IR spectra are shown in Fig. 1c. As shown in Fig. 1c, the FT-IR spectrum of the obtained Fe–Ce/MCM-41 was in good agreement with that of MCM-41 except for a slight shift in the position. The Si–O–Si bands at 1090 cm−1 and 801 cm−1 were caused by asymmetrical stretching and symmetrical stretching, respectively. The strong absorption at 960 cm−1 and 461 cm−1 was assigned to Si–O stretching and twisting, respectively.36 The results suggested that the framework was not destroyed after the loading of iron and cerium oxides onto the MCM-41 surface.
To investigate the composition and chemical state of Fe–Ce/MCM-41 before and after the reaction, XPS analysis was carried out. As shown in Fig. 1d, the major composition of fresh Fe–Ce/MCM-41 and aged Fe–Ce/MCM-41 catalysts consists of iron (Fe), cerium (Ce), oxygen (O) and carbon (C). The atomic ratios of C, O, Fe and Ce before and after the reaction are shown in the Fig. 1d inset. The atomic percentage of Fe decreased from 5.77% to 4.31%, however, Ce only decreased slightly, which can be neglected compared to that of Fe.
The nitrogen adsorption/desorption isotherms of MCM-41 and Fe–Ce/MCM-41 (Fig. 1e) correspond to type IV isotherms, which indicates that they possess a mesoporous structure. The BJH desorption pore size distribution is shown in Fig. 1f, and this distribution indicates that both MCM-41 and Fe–Ce/MCM-41 have a centralized distribution in a 2–4 nm range, which further confirms that the obtained products were primarily mesoporous. The detailed texture parameters of MCM-41 and Fe–Ce/MCM-41 are shown in Table S1.† Despite the slight decrease in the average pore size, Fe–Ce/MCM-41 retains a relatively large surface area and pore volume, which could provide more active sites for the decomposition of MO in the presence of H2O2.
Fig. 2 shows the HRTEM micrographs of MCM-41 and Fe–Ce/MCM-41. As shown in Fig. 2b, the obtained products exhibited an ordered mesoporous structure that was identical to MCM-41.30 Although a slight agglomeration phenomenon was observed on the surface of Fe–Ce/MCM-41, the ordered mesoporous structure was not destroyed. The slight agglomeration phenomenon also indicated that the iron and cerium oxides were uniformly dispersed on the surface of Fe–Ce/MCM-41.
 | ||
| Fig. 2 HRTEM images of MCM-41 (a) and Fe–Ce/MCM-41 (b). | ||
3.2 Degradation of MO using the Fe–Ce/MCM-41 catalyst
Prior to testing the catalytic ability of mesoporous MCM-41, Fe–MCM-41 and Fe–Ce/MCM-41, the effects of adsorption processes were evaluated in the absence H2O2. As shown in Fig. 3a, only 9.1%, 10.2% and 10.6% MO were removed by MCM-41, Fe–MCM-41 and Fe–Ce/MCM-41, respectively. The slight increase in the adsorption of Fe–Ce/MCM-41 compared to that of Fe–MCM-41 may be due to the larger surface area of Fe–Ce/MCM-41, which promoted surface adsorption of MO. | ||
| Fig. 3 (a) MO removal by adsorption on MCM-41, Fe–MCM-41 and Fe–Ce/MCM-41. Experimental conditions: pH 5.0, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO. (b) Removal of MO in various catalytic processes. Experimental conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO. | ||
In the presence of H2O2, the catalytic activities of various processes were evaluated. The degradation curves that are shown in Fig. 3b indicate that H2O2 only results in minor removal of MO, and Ce–MCM-41 leads to MO degradation but the efficiency was relatively low, which may be due to surface adsorption. Using mesoporous Fe–Ce/MCM-41 as the heterogeneous catalyst, the removal efficiency was notably higher than that with Fe–MCM-41, and the removal efficiencies were 99.5 and 76%, respectively, enhancing the mass transfer and chemical reaction at the reactive sites. Therefore, the large surface area of Fe–Ce/MCM-41 provides more reactive sites. In addition, the catalytic activity was enhanced due to the incorporation of cerium. In comparison, the catalytic activity of the Fe–Ce/MCM-41 composite was higher than that of the Fe/MCM-41 and Ce/MCM-41 mixture, suggesting that a synergetic effect may exist in the composite, which enhances the chemical reaction at the reactive sites.
3.3 Effects of different operating conditions on MO degradation
Due to the solution pH being a major factor in the Fenton reaction, the effects of pH value on the removal of MO solution were investigated, and the results are shown in Fig. 4a. These results indicate that MO degradation decreased as the pH increased, and the best removal efficiency was obtained in a pH range of 3.0–5.0. In comparison to the homogeneous Fenton process, Fe–Ce/MCM-41 broadened the pH range, which decreased the costs of acidification during processing and neutralization after treatment. Additionally, the degradation rate constants (Fig. 4a) at pH values of 3.0 and 5.0 were 0.093 and 0.083 min−1, respectively, which were higher than those previously reported by Wen.37 In addition, the effects of the pH of the MO solution on the leaching of iron was also invested (Fig. S2†). Based on our results, additional research will be carried out at a pH of 5.0. | ||
| Fig. 4 Different operating conditions for MO degradation: (a) initial pH value, (b) H2O2 dosage, (c) Fe–Ce/MCM-41 amount, and (d) MO concentration. Except for the investigated parameter, other parameters were fixed: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO. | ||
Fig. 4b shows the impact of different H2O2 concentrations on the degradation of the MO solution by the Fe–Ce/MCM-41 Fenton-like systems. MO removal at different H2O2 concentrations followed the first-order-rate law, and the observed degradation rate constants increased from 0.014 to 0.083 min−1 as the H2O2 dosage increased from 10 to 20 mM. A previous study38 reported that an appropriate H2O2 concentration can increase the amount of ˙OH radicals that are formed. However, with the excess addition of H2O2, the k value decreased to 0.023 min−1, which may be related to the scavenging effect of ˙OH radicals according to eqn (1) and (2).
| H2O2 + ˙OH → H2O + ˙OOH | (1) |
| ˙OOH + ˙OH → H2O + O2 | (2) |
The kinetics of the MO degradation were also studied with different catalyst dosages (1.0, 2.0, 3.0, and 4.0 g L−1) (Fig. 4c). As the addition amount of the Fe–Ce/MCM-41 catalyst increased from 1.0 to 4.0 g L−1, the rate constant increased and then slightly decreased. The increase in the removal efficiency may be due to the increasing formation of ˙OH, and the slight decrease in the removal efficiency (Fig. S3†) may be due to the self-scavenging effect of ˙OH by excess Fe2+, according to eqn (3) and (4).39
| Fe2+ + ˙OH → Fe3+ + OH− | (3) |
| Fe2+ + ˙OOH → Fe3+ + OOH− | (4) |
The influence of different initial MO concentrations on the degradation progress was also investigated. The results (Fig. 4d) indicated that the degradation rate decreased as the MO concentration increased from 50 to 200 mg L−1, which may be due to an increase in the MO concentration competing with the number of HO˙ radicals in the solution. It is important to note that a high removal efficiency can be obtained at a high MO concentration (Fig. S4†). However, the rate constants decreased from 0.095 to 0.039 min−1 as the MO concentration increased from 50 to 200 mg L−1. This case revealed that the removal rate of MO was inhibited by the high concentration of the MO solution in the Fe–Ce/MCM-41 Fenton-like system, which may be due to the limited generation rate of ˙OH radicals in the solution.
Previous studies have reported that the solubility and dissolution rate of a mineral are sensitive to pH, and a higher solubility and faster dissolution have been observed in neutral and alkaline solutions.40,41 Pham et al.42 reported that mesoporous silica materials, such as SBA-15, HMS and MCM-41, can change the reactivity of catalysts in pH 7–10 solutions. In the current study, the experiment was performed at a pH of 5. Therefore, the silica can be slowly dissolved, which will have a smaller effect on the catalytic activity of the Fe–Ce/MCM-41 composite.
3.4 Possible degradation mechanism of MO
To determine the contribution of homogeneous Fenton reaction that is catalysed by the leached Fe and Ce ions on MO degradation, the concentrations of dissolved iron and cerium ions as well as the MO removal efficiency were investigated under following conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, temperature 30 °C and 100 mg L−1 MO, and the results are shown in Fig. 5a. The total dissolved iron continuously increased due to leaching of ferrous ions and ferric ions from the Fe–Ce/MCM-41 catalyst. After 120 min, the loss of iron was as low as 0.73 mg L−1, implying that the main iron species were retained on the Fe–Ce/MCM-41 catalyst. No dissolved cerium was detected during the entire experiment. Then, the homogeneous experiment was performed by removing the Fe–Ce/MCM-41 catalyst after vigorous agitation for 120 min, followed by the addition of 224 μL H2O2 (20 mM) to initiate the reaction. As shown in Fig. 3a, the removal of MO due to adsorption was 9%, and the removal of MO at 120 min was 24% (Fig. 5a) due to the homogeneous Fenton reaction (catalysed by the leached Fe ions). Therefore, the contribution of the homogeneous Fenton-like reaction was only approximately 13%. Nevertheless, the oxidation of MO by the Fe–Ce/MCM-41 catalyst was nearly 100% at 120 min. These results indicated that the contribution from the homogeneous reaction was minor, and the degradation of MO was primarily dominated by the heterogeneous reaction.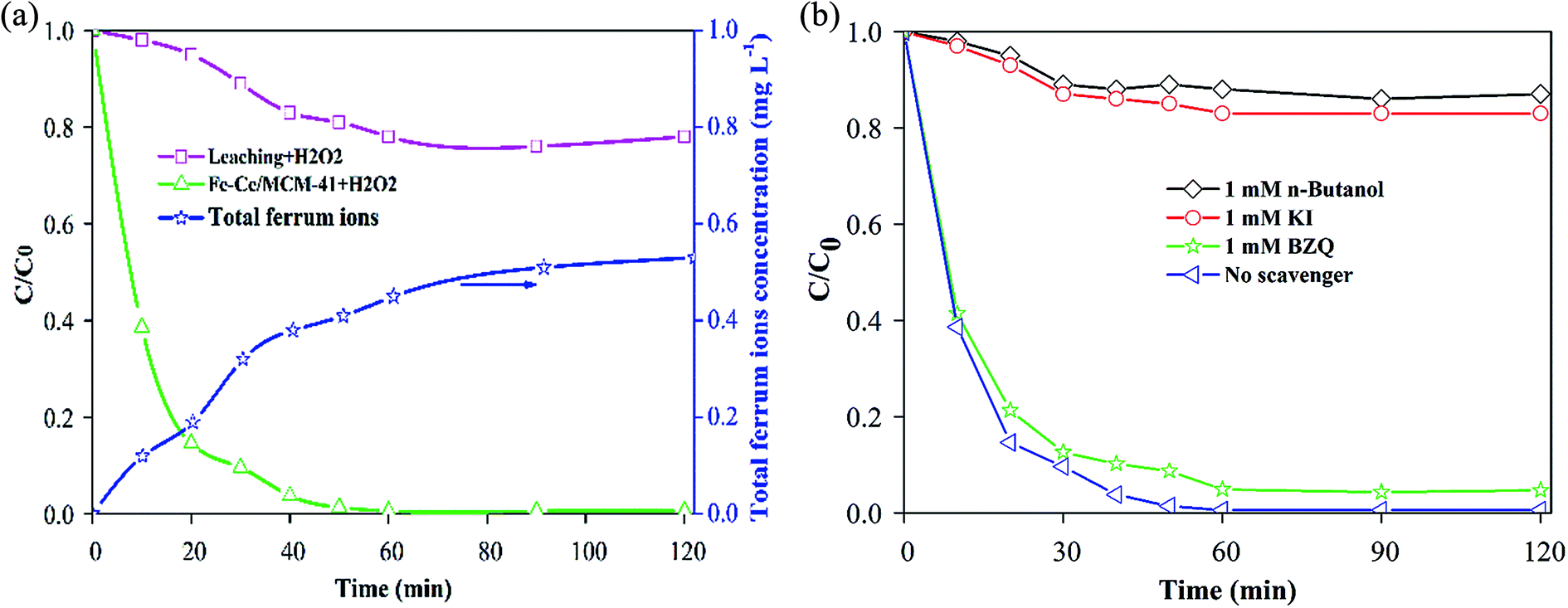 | ||
| Fig. 5 (a) MO removal efficiency and concentration of total ferrum ions (operating conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO). (b) Degradation efficiency of MO with different scavengers. | ||
The reactive species are the main participants in pollutant degradation processes. Therefore, n-butanol was used as an OH˙ scavenger, KI was used as a surface OH˙ scavenger (˙OHads), and benzoquinone (BZQ) was used as a superoxide radical (O2˙) scavenger, and these scavengers were introduced into the oxidation system to determine the dominant species in the MO degradation process. As shown in Fig. 5b, the catalytic activity of Fe–Ce/MCM-41 decreased from 99.6% to 13.1%, when 1 mM OH˙ scavenger (n-butanol) was added to the reaction system. In the presence of excess KI (1 mM), the removal efficiency of MO decreased substantially to 17.3%, indicating that ˙OHads play a dominant role in promoting rapid oxidation of MO. When 1 mM O2˙ scavenger (BZQ) was added, the removal efficiency of MO by the Fe–Ce/MCM-41 catalyst was approximately 95.3%. These results indicate that the dominant active species was ˙OHads.
Fig. 6a shows the Fe 2p spectra of the fresh and aged Fe–Ce/MCM-41 catalysts. The binding energies at 724.6 eV and 711.3 eV correspond to Fe 2p1/2 and Fe 2p3/2, respectively. Additionally, the Fe 2p3/2 peak can be divided into two components with binding energies of 710.8 eV and 712.9 eV, indicating the presence of Fe2+ and Fe3+, respectively.24 The results of the fresh and aged Fe–Ce/MCM-41 indicated that the area of Fe3+ increased at a binding energy of 712.9 (from 48.31% to 52.25%), which may be due to the oxidation of Fe2+ and Fe3+ on the surface of Fe–Ce/MCM-41.
 | ||
| Fig. 6 (a) Fe 2p spectra of fresh Fe–Ce/MCM-41 and aged Fe–Ce/MCM-41. (b) Ce 3d spectra of fresh Fe–Ce/MCM-41 and aged Fe–Ce/MCM-41. (c) O 1s spectra of fresh Fe–Ce/MCM-41 and aged Fe–Ce/MCM-41 (experimental conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO). | ||
The Ce 3d XPS spectra of Fe–Ce/MCM-41 before and after the reaction are shown in Fig. 6b. Based on previous studies, the XPS spectra of Ce 3d were characterized by complex but distinct features that were associated with the final-state occupation of the Ce 4f level.32 The hybridization results split the peaks into doublets, which are referred to as u/v, u′/v′, u′′/v′′ and u′′′/v′′′, where u and v correspond to the 3d3/2 and 3d5/2 spin–orbit states, respectively. The u/v, u′′/v′′ and u′′′/v′′′ doublets are due to the 3d104f0 Ce4+ final state, and the u′/v′ doublet was due to the 3d104f1 final state, corresponding to Ce3+. For the fresh Fe–Ce/MCM-41, the total areas of the u/v, u′′/v′′ and u′′′/v′′′ peaks were much higher than that of the u′/v′ peak, indicating that Ce4+ is the main Ce species in the Fe–Ce/MCM-41 catalyst. However, after reaction for 120 min, the areas of the u/v, u′′/v′′ and u′′′/v′′′ peaks decreased, and in particular, the area of the u′′′/v′′′ peak decreased substantially (from 41.64% to 28.29%), indicating that the Ce4+ content decreased. The reduction of Ce4+ to Ce3+ may be due to electrons being transferred from ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe2+ to Ce4+, where Fe2+ and Ce4+ represent the Fe2+ and Ce4+ sites, respectively, on the catalyst surface. Due to the standard redox potentials of Ce4+/Ce3+ and Fe3+/Fe2+, which are 1.44 V and 0.77 V, respectively, the transfer process from Fe2+ to Ce4+ is thermodynamically favoured.33 Therefore, the coexistence of Ce4+/Ce3+ and Fe3+/Fe2+ plays an important role in promoting the heterogeneous Fenton-like reaction.
Fe2+ to Ce4+, where Fe2+ and Ce4+ represent the Fe2+ and Ce4+ sites, respectively, on the catalyst surface. Due to the standard redox potentials of Ce4+/Ce3+ and Fe3+/Fe2+, which are 1.44 V and 0.77 V, respectively, the transfer process from Fe2+ to Ce4+ is thermodynamically favoured.33 Therefore, the coexistence of Ce4+/Ce3+ and Fe3+/Fe2+ plays an important role in promoting the heterogeneous Fenton-like reaction.
Fig. 6c shows the O 1s spectra of the Fe–Ce/MCM-41 catalyst before and after the reaction. As shown in Fig. 6c, two types of oxygen were present in the catalyst (i.e., the lattice oxygen (O1) and chemisorbed oxygen (O2)).32 The O 1s peaks at approximately 528.9–533.2 eV were due to the lattice oxygen of SiO2, and the peaks at approximately 534.1–534.6 eV were due to chemisorbed oxygen of Fe3O4, Fe2O3 and CeO2. The chemisorbed oxygen was more active than the lattice oxygen, which played an important role in the oxidation process.
Based on the aforementioned analyses, both surface-bound ˙OH radicals (˙OHads) and free ˙OH radicals (˙OHfree) play significant roles in the oxidation of MO. Additionally, a possible reaction mechanism for MO oxidation has been proposed in Fig. 7. The reaction was initiated on the catalyst surface, and Fe2+ reacted with H2O2 to generate ˙OHads, as shown in eqn (5). As a reversible reaction, Fe2+ was regenerated by reaction of the formed Fe3+, according to eqn (6), and Fe3+ on the surface of Fe–Ce/MCM-41 with H2O2 (eqn (7)), which was important for the continuous reaction. The oxidation chains (eqn (5)–(7)) could also be applied for the cerium species, and the reaction process is shown in (eqn (8) and (9)), as reported by Xu.39 Moreover, the transfer of electrons from Fe2+ to Ce4+ was thermodynamically possible because the standard redox potentials of Ce4+/Ce3+ and Fe3+/Fe2+ are 1.44 V and 0.77 V, respectively (eqn (10)). After the dissolved iron was dispersed into the bulk solution from the surface of the Fe–Ce/MCM-41 catalyst, a series of reactions occurred (eqn (11)–(13)). An excess amount of H2O2 and Fe2+ resulted in negative effects on the degradation of MO, as demonstrated in 3.2 (eqn (1)–(4)). Finally, MO was primarily oxidized by ˙OHads/free in the bulk solution.
|
Fe2+ + H2O2 + H+ → Fe3+ + ˙OHads + H2O
| (5) |
|
Fe3+ + H2O2 → Fe2+ + HO2˙ + H+
| (6) |
|
Fe3+ + HO2˙ → Fe2+ + O2 + H+
| (7) |
|
Ce3+ + H2O2 + H+ → Ce4+ + ˙OHads + H2O
| (8) |
|
Ce4+ + HO2˙ → Ce3+ + O2
| (9) |
|
Fe2+ + Ce4+ ↔ Fe3+ + Ce3+
| (10) |
| Fe2+ + H2O2 + H+ → Fe3+ + ˙OHfree + H2O | (11) |
| Fe3+ + H2O2 → Fe2+ + HO2˙ + H+ | (12) |
| Fe3+ + HO2˙ → Fe2+ + O2 + H+ | (13) |
| ˙OHads/free + MO → intermediates | (14) |
| ˙OHads/free + intermediates → CO2 + H2O | (15) |
 | ||
| Fig. 7 Reaction mechanism of H2O2 activation by the Fe–Ce/MCM-41 catalyst. | ||
3.5 Mineralization ability and reusability of the catalyst
The TOC is an important indicator of water contamination.43 Therefore, the degree of MO mineralization is shown in Fig. 8a. The TOC removal was 53% with a 60 min reaction, and the maximum TOC removal reached 65% after a 120 min reaction. These results indicated that MO can be oxidized to H2O and CO2 via the Fe–Ce/MCM-41 catalysed H2O2 system. | ||
| Fig. 8 (a) Temporal change in MO and TOC removal. (b) Repeated use of Fe–Ce/MCM-41 for MO removal in a bulk solution (experimental conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 Fe–Ce/MCM-41 catalyst, T = 30 °C and 100 mg L−1 MO). | ||
Because the reusability of the catalyst is important for practical applications, successive batch experiments of MO degradation were carried out. As shown in Fig. 8b, the activity of the Fe–Ce/MCM-41 catalyst decreased little during the five runs after the 60 min reaction. This phenomenon confirmed the excellent stability of the Fe–Ce/MCM-41 catalyst.
4. Conclusions
A simple and active Fe–Ce/MCM-41 catalyst with a highly ordered mesoporous structure, large surface area, and high pore volume was synthesized via a facile ultrasonic dispersion method. The Fe–Ce/MCM-41 catalyst with a loading concentration of 7.5 wt% exhibited excellent catalytic activity for the oxidation of MO in the dark, and a removal efficiency of nearly 100% was achieved after 60 min with the following reaction conditions: pH 5.0, 20 mM H2O2, 2.0 g L−1 catalyst, temperature 30 °C and 100 mg L−1 MO solution. Based on the homogeneous experiment and scavenger analysis results, MO in the bulk solution was primarily oxidized by surface-bound ˙OH radicals (˙OHads) and free ˙OH radicals (˙OHfree), and in particular, ˙OHads played a dominant role. In addition, excellent mineralization ability and superior stability of the Fe–Ce/MCM-41 catalyst were observed. We believe that the results from this study provide an efficient Fenton-like catalyst and pave the way for the potential applications.Acknowledgements
This work was financially supported by the National Science and Technology Major Project (No. 2016ZX05040003) and Postgraduate Innovation Project of China University of Petroleum (No. YCX2015032).References
- C. Chang, Y. Fu, M. Hu, C. Wang, G. Shan and L. Zhu, Appl. Catal., B, 2013, 142–143, 553–560 CrossRef CAS.
- C. Qi, X. Liu, C. Lin, X. Zhang, J. Ma, H. Tan and W. Ye, Chem. Eng. J., 2014, 249, 6–14 CrossRef CAS.
- H. Sun, C. Kwan, A. Suvorova, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2014, 154–155, 134–141 CrossRef CAS.
- R. Liu, Y. Guo, Z. Wang and J. Liu, Chemosphere, 2014, 95, 643–646 CrossRef CAS PubMed.
- B. Jiang, J. Zheng, X. Lu, Q. Liu, M. Wu, Z. Yan, S. Qiu, Q. Xue, Z. Wei, H. Xiao and M. Liu, Chem. Eng. J., 2013, 215–216, 969–978 CrossRef CAS.
- W. A. Al-Amrani, P.-E. Lim, C.-E. Seng and W. S. Wan Ngah, J. Taiwan Inst. Chem. Eng., 2014, 45, 609–616 CrossRef CAS.
- M. A. Rauf, M. A. Meetani and S. Hisaindee, Desalination, 2011, 276, 13–27 CrossRef CAS.
- P. Jing, J. Li, L. Pan, J. Wang, X. Sun and Q. Liu, J. Hazard. Mater., 2015, 284, 163–170 CrossRef CAS PubMed.
- Z. Xiong, B. Lai, P. Yang, Y. Zhou, J. Wang and S. Fang, J. Hazard. Mater., 2015, 297, 261–268 CrossRef CAS PubMed.
- X. Zhang, X. B. Wang, L. W. Wang, W. K. Wang, L. L. Long, W. W. Li and H. Q. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 7766–7772 CAS.
- W. H. Abdelraheem, X. He, X. Duan and D. D. Dionysiou, J. Hazard. Mater., 2015, 282, 233–240 CrossRef CAS PubMed.
- Y. Lei, C. S. Chen, Y. J. Tu, Y. H. Huang and H. Zhang, Environ. Sci. Technol., 2015, 49, 6838–6845 CrossRef CAS PubMed.
- B. Ileri, O. Ayyildiz and O. Apaydin, J. Hazard. Mater., 2015, 292, 1–8 CrossRef CAS PubMed.
- R. Liu, D. Xiao, Y. Guo, Z. Wang and J. Liu, RSC Adv., 2014, 4, 12958 RSC.
- B. Jiang, P. Hu, X. Zheng, J. Zheng, M. Tan, M. Wu and Q. Xue, Chemosphere, 2015, 125, 220–226 CrossRef CAS PubMed.
- Z. Wang, R. T. Bush, L. A. Sullivan, C. Chen and J. Liu, Environ. Sci. Technol., 2014, 48, 3978–3985 CrossRef CAS PubMed.
- J. Lu, J. Wu, Y. Ji and D. Kong, Water Res., 2015, 78, 1–8 CrossRef CAS PubMed.
- C. Qi, X. Liu, W. Zhao, C. Lin, J. Ma, W. Shi, Q. Sun and H. Xiao, Environ. Sci. Pollut. Res. Int., 2015, 22, 4670–4679 CrossRef CAS PubMed.
- Z. Wang, W. Ma, C. Chen and J. Zhao, J. Hazard. Mater., 2009, 168, 1246–1252 CrossRef CAS PubMed.
- B. Jiang, X. Wang, Y. Liu, Z. Wang, J. Zheng and M. Wu, J. Hazard. Mater., 2015, 304, 457–466 CrossRef PubMed.
- L. Fu, Z. Zhao, J. Ma and X. Hu, Catal. Commun., 2015, 65, 96–101 CrossRef CAS.
- Z. Wang, RSC Adv., 2014, 4, 31476 RSC.
- X. Hu, B. Liu, Y. Deng, H. Chen, S. Luo, C. Sun, P. Yang and S. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef CAS.
- T. D. Nguyen, N. H. Phan, M. H. Do and K. T. Ngo, J. Hazard. Mater., 2011, 185, 653–661 CrossRef CAS PubMed.
- F. Qi, W. Chu and B. Xu, Chem. Eng. J., 2015, 262, 552–562 CrossRef CAS.
- F. Qi, B. Xu and W. Chu, J. Mol. Catal. A: Chem., 2015, 396, 164–173 CrossRef CAS.
- Z. H. Wang, D. X. Xiao, R. L. Liu, Y. G. Guo, X. Y. Lou and J. S. Liu, J. Adv. Oxid. Technol., 2014, 17, 104–108 CAS.
- M. A. G. Hinkle, Z. Wang, D. E. Giammar and J. G. Catalano, Geochim. Cosmochim. Acta, 2015, 158, 130–146 CrossRef CAS.
- S. Song, J. Tu, L. Xu, X. Xu, Z. He, J. Qiu, J. Ni and J. Chen, Chemosphere, 2008, 73, 1401–1406 CrossRef CAS PubMed.
- Z. Ying, Z. Li, Y. Zheng, X. Shen and L. Lin, Mater. Lett., 2006, 60, 3221–3223 CrossRef CAS.
- P. Horcajada, A. Rámila, J. Pérez-Pariente and M. Vallet-Regí, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 5162–5167 CrossRef CAS.
- L. Xu and J. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CAS.
- Z. Xiong, B. Lai, P. Yang, Y. Zhou, J. Wang and S. Fang, J. Hazard. Mater., 2015, 297, 261–268 CrossRef CAS PubMed.
- P. Feng, X. Guan, Y. Sun, W. Choi, H. Qin, J. Wang, J. Qiao and L. Li, J. Environ. Sci., 2015, 31, 175–183 CrossRef PubMed.
- B. Li, W. Ma, C. Han, J. Liu, X. Pang and X. Gao, Microporous Mesoporous Mater., 2012, 156, 73–79 CrossRef CAS.
- Z. Wen, Y. Zhang, C. Dai and Z. Sun, J. Hazard. Mater., 2015, 287, 225–233 CrossRef CAS PubMed.
- S. Fukuchi, R. Nishimoto, M. Fukushima and Q. Zhu, Appl. Catal., B, 2014, 147, 411–419 CrossRef CAS.
- L. Xu and J. Wang, J. Hazard. Mater., 2011, 186, 256–264 CrossRef CAS PubMed.
- C. P. G. A. E. J. Reardon, J. Phys. Chem. A, 2008, 112, 3386–3390 CrossRef PubMed.
- M. N. A. Galarneau, F. Guenneau, F. Di Renzo and A. Gedeon, J. Phys. Chem. C, 2007, 111, 8268–8277 Search PubMed.
- A. L. Pham, D. L. Sedlak and F. M. Doyle, Appl. Catal., B, 2012, 126, 258–264 CrossRef CAS PubMed.
- P. Hu, Y. Liu, B. Jiang, X. Zheng, J. Zheng and M. Wu, Ind. Eng. Chem. Res., 2015, 54, 8277–8286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26820f |
| This journal is © The Royal Society of Chemistry 2016 |