
Density functional theory study of the mechanism of a dipeptide-catalyzed intermolecular aldol reaction—the effects of steric repulsion interactions on stereoselectivity†
Xiaofei Zhangab and
Min Pu*a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: pumin@mail.buct.edu.cn
bInner Mongolia Key Laboratory of Photoelectric Functional Materials, Chifeng University, Chifeng, 024000, China
First published on 9th February 2016
Abstract
The mechanism of the dipeptide (S)-pro-(S)-asp catalyzed intermolecular aldol reaction with acetone as the donor and three aromatic aldehydes (benzaldehyde, p-methyl benzaldehyde and p-nitrobenzaldehyde) as the acceptors was studied by means of density functional theory (DFT) at the level of B3LYP/6-31G(d,p). The calculated results showed that there were four steps in the reaction path: (i) the nucleophilic attack of an amino group on carbonyl for the formation of intermediate A, which was the rate-determining step due to it having the largest energy barrier of 44.33 kcal mol−1; (ii) the dehydration process to form an s-cis- or s-trans-enamine through an imine-generating step; (iii) the electrophilic addition of aldehyde, which decided the stereoselectivity of the product because of the steric repulsion interactions between the enamine and aldehyde; (iv) the removal of the dipeptide to generate the final products. According to the results analysis, it was found that the dipeptide-catalyzed aldol reaction via an s-trans-enamine was more energetically favorable to obtain the R-product (with an ee value > 99%). The energy variations in the reaction path were verified using CAM-B3LYP and M06-2X methods in the same basis set. The solvation effects were explored based on B3LYP/6-31G(d,p) combined with a polarizable continuum model (PCM), the substituent effects of aromatic aldehydes were also considered. The computed results provided a reference for experiments that DMSO and H2O as the solvents could decrease the energy barriers in the reaction path and the impact of substituent effects might be small. The feasibility of the dipeptide provided a possibility for proteins to act as catalysts which are green and nontoxic.
1. Introduction
A series of peptides and amino acids used as catalysts for the asymmetric intermolecular aldol reaction which was an important C–C bond-forming reaction in the synthesis of organic products and pharmaceuticals had been studied and attracted much attention.1–13 L-Proline-based small peptides were developed as efficient catalysts for the asymmetric direct aldol reactions of acetone with aldehydes, and high yields and stereoselectivity of up to 96% ee were obtained in aqueous media.14 The subsequent research approved that small di- to tetra-peptides with a primary amine functionality also catalyzed the asymmetric intermolecular aldol reaction with excellent enantioselectivity.15,16 In addition, the small peptides could catalyze the aldol reaction in water and non-aqueous media with a high yield asymmetric product.17 Córdova et al. investigated the origins of the stereoselectivity for the asymmetric intermolecular aldol reaction using DFT calculations.18,19 The results showed that amino acids with a primary amino functionality were able to catalyze this kind of reaction with high stereoselectivity.Heine and Wong et al.20,21 researched the enzymatic-catalyzed asymmetric aldol reaction and concluded that the reaction involved two classes of intermediates, Type I aldolases (enamine intermediates) and Type II aldolases (Zinc enolate intermediates). In Type I aldolases, the primary amino group of the amino acid residue had a large effect on the reaction process, in which enamine intermediates formed with the help of proton transfer by neighboring amino acids. In Type II aldolases, chiral enolates, chiral aldehydes, or chiral auxiliaries were used to control the stereoselectivity of the aldol reactions.
In 1974, Hajos et al.22 found that amino acids were able to catalyze the asymmetric aldol reaction via an enamine intermediate path. Nevertheless, it was not rapidly developed until List et al.23,24 demonstrated that proline and its derivatives were highly enantioselective catalysts for the intermolecular asymmetric aldol reaction between ketones and aldehydes in 2000. From then on, amino acids were used more often as highly selective catalysts for intermolecular aldol reactions.25,26 Generally speaking, this kind of proline-catalyzed reaction involves an enamine-forming process where one proline molecule took part in the transition state as suggested by Houk and List.27–30 A lot of work to get highly efficient and stereoselective asymmetric catalysts has been carried out since that time, in which the catalysis of proline derivatives and the expansion of the scope of substrates were widely researched.31–33 The high dr (up to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ee values (up to 98%) of the targeted product were obtained using primary amine-containing amino acids as catalysts, such as L-threonine and O-tBu-L-tyrosine.34
:1) and ee values (up to 98%) of the targeted product were obtained using primary amine-containing amino acids as catalysts, such as L-threonine and O-tBu-L-tyrosine.34
Recently, small peptides were also suggested as effective catalysts for the asymmetric aldol reaction.35 Isolated from living cells in vitro, these peptides could catalyze the asymmetric aldol reaction, demonstrating that non-enzymatic enantioselective catalysis could occur in living cells and be of biological relevance.17 The beneficial effect of the small peptide catalyzed asymmetric aldol reaction was due to the faster hydrolysis of the intermediates in the enamine catalytic cycle, as well as the suppression of non-productive imidazolidinone formation.15 However, the mechanism of the small peptide catalyzed intermolecular aldol reaction was not distinct for the moment.
Proline typically gave superior results of yield and enantioselectivity in the above mentioned catalytic studies, and even promoted the finding of several other catalysts. To the best of our knowledge, there are few theoretical investigations concerning the detailed variation of the dipeptide-catalyzed aldol reactions between ketones and aromatic aldehydes, although great efforts have been made to explain the stereoselectivity of the reactions. Accordingly, the mechanism and stereoselectivity of the intermolecular aldol reaction catalyzed by a dipeptide was researched and the transition states of the C–C bond formation step were found using density functional theory methods in this paper. The calculations successfully predicted the stereoselectivity of the observed product and provided the key insights into the detailed reaction paths.
2. Computational methods
All the calculations were performed using the gradient-corrected function B3LYP of Density Functional Theory (DFT). The traditional hybrid Becke, three-parameter, Lee–Yang–Parr (B3LYP) exchange correlation functions, the long-range-corrected Coulomb-attenuating method functional (CAM-B3LYP) and hybrid meta-generalized gradient approximation functional (M06-2X) of DFT were applied. The geometries were optimized using the standard double zeta plus polarization basis set 6-31G(d,p). Meanwhile, the single point energies of species in the gas phase were tested and verified at the level of B3LYP/6-311++G(d,p) which is listed in the ESI.† Frequency calculations were performed at the same theory level as the optimizations to obtain zero-point energies (ZPEs) and to confirm the nature of the stationary points. To investigate the transition states to the two desired minima of the proposed mechanism, the potential energy profile was generated by using intrinsic reaction coordinate (IRC) calculations. The solvent effects on the mechanism were discussed based on the polarizable continuum model (PCM) of self-consistent reaction field (SCRF) theory. All of the quantum chemical calculations were applied using Gaussian 09 programs.363. Results and discussion
3.1 Dipeptide structure
The dipeptide, perhaps, functioned as a “micro-aldolase” that provided both the nucleophilic amino group and electrophilic carbonyl cocatalyst in the form of the carboxylate. To investigate the mechanism of the dipeptide-catalyzed intermolecular aldol reaction, a model with acetone as the donor and benzaldehyde as the acceptor was chosen to study the (S)-pro-(S)-asp-catalyzed aldol reaction. The catalyst (S)-pro-(S)-asp was formed by the dehydration of (S)-proline and (S)-aspartic acid. As shown in Fig. 1, the four atoms of the peptide bond and its attached C atoms of both ends were in the same plane. The peptide bond was a stable covalent bond because the bonding of C–N was sp2 hybridised in which the conjugation structure was subsistent. | ||
| Fig. 1 Optimized structure of dipeptide (S)-pro-(S)-asp. | ||
3.2 Reaction mechanism
The most possible mechanism of the dipeptide-catalyzed intermolecular aldol reaction is depicted in Fig. 2. The proposed mechanism contained four steps: (i) the nucleophilic attack on the carbonyl group by the amino group for the formation of intermediate A; (ii) the dehydration process to form an s-cis- or s-trans-enamine; (iii) the electrophilic addition of the aldehyde; (iv) the deprivation of catalyst-dipeptide to the final products. The discussions will be carried out in detail and step by step in the following parts. Pioneering theoretical studies had determined that the aldol reactions proceed via enamine intermediates, while the transition states for the crucial C–C bond-forming step (nucleophilic addition of the enamine intermediate to an electrophilically activate aldehyde) showed that an arrangement of the reacting atoms was stabilized by a hydrogen-bonding interaction between the proton of the amino-group moiety in the peptide bond and the oxygen atom of the electrophile.37–39 However, the stereoselectivity of the products was determined by the spatial orientation and steric repulsion of groups in transition states. | ||
| Fig. 2 Reaction mechanism of the dipeptide-catalyzed aldol reaction. | ||
 | ||
| Fig. 3 Optimized structure of transition state TS1. | ||
 | ||
| Fig. 4 Formation of two enamines by three paths. | ||
The other probable path (N-path) was that the hydroxyl firstly reacted with the H(7) atom on the amino group of the peptide bond to remove the H2O molecule (transition state of dehydration in N-path in Fig. 5(a)) and form an imine simultaneously, then a hydrogen atom of either methyl was transferred to the N(4) atom to form the s-cis- or s-trans-isomer. The corresponding first energy barrier of imine forming was 13.35 kcal mol−1, and the next energy barriers were 8.26 and 19.35 kcal mol−1 for the s-cis- and s-trans-enamines, respectively, which revealed that the s-cis-enamine was more energetically favorable and occurred easily. The calculated conversion energy barrier of the two enamines was 5.06 kcal mol−1 indicating that the s-cis-enamine was achieved mostly during the proton transfer process, and then the s-trans-enamine was obtained through configuration conversion from the s-cis-enamine in the N-path. This path was a facile process and attained easily under the experimental conditions. Besides, there was a potential path (O-path) that the hydroxyl firstly reacted with the H(8) atom on the terminal carboxylic acid group of the dipeptide to remove the H2O molecule (transition state of dehydration in O-path in Fig. 5(b)) and formed an imine simultaneously, then the hydrogen atom of either methyl transferred to the O(5) atom to form the s-cis- or s-trans-isomer. The corresponding first energy barrier of imine forming was 13.36 kcal mol−1, and the next energy barriers were 1.86 and 1.69 kcal mol−1 for the s-cis- and s-trans-enamines, respectively, which demonstrated that the two isomers were almost achieved in a 1:1 ratio and performed easily at room temperature. The energy variations in the three paths to form two enamines are shown in Fig. 6.
 | ||
| Fig. 5 Transition states of dehydration process in N-path and O-path. | ||
 | ||
| Fig. 6 The energy variations in three paths to form enamines. | ||
The energy barriers of the proton transfer forming an enamine in the O-path were smaller than that of the N-path, it might be because a proton transferred from the imine to the carboxylic group could more easily overcome the steric repulsion and bone torsion force. In summary, the O-path was preferential to generate half-to-half s-cis- and s-trans-enamines in the second step from the perspective of energy. Therefore, the subsequent discussion of the mechanism, energy variation and solvent effects was carried out around the favorable O-path.
The structures of two enamines are shown in Fig. 7 (the detailed geometric parameters of enamines are listed in the ESI†).
 | ||
| Fig. 7 Structural models of two enamines. | ||
 | ||
| Fig. 8 Transition states in third step. | ||
The aldol reaction was proposed involving an enamine intermediate, and the stereo-controlling step was the formation of a C–C bond between the enamine intermediate and benzaldehyde. The computed energies of the transition structures associated with the C–C bond-formation step could reasonably explain the stereoselectivity of the intermolecular aldol reaction.
The energetically most accessible transition state, TS3-tR, had several features that made it have lower energy than all the other ones in the gas phase. The configuration of the dipeptide was optimal to form a hydrogen bond (N–H⋯O) between the dipeptide amide and the carbonyl that was developing on benzaldehyde. The amide moiety was also in an optimum position to donate its proton and prevent steric repulsion from the framework. In addition, the developing anion of the benzaldehyde interacted favorably with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moiety of the s-trans-enamine. These interactions stabilized the TS3-tR to a very high degree, making the concerted C–C bond formation and proton transfer from the amide group very synchronous. Furthermore, the aspartic acid substituent of the dipeptide was pointing away from benzaldehyde and at the same time avoiding steric repulsion from the dipeptide backbone.
C moiety of the s-trans-enamine. These interactions stabilized the TS3-tR to a very high degree, making the concerted C–C bond formation and proton transfer from the amide group very synchronous. Furthermore, the aspartic acid substituent of the dipeptide was pointing away from benzaldehyde and at the same time avoiding steric repulsion from the dipeptide backbone.
It should be worth noting that there were two possible stereoselective channels including the cis-isomer and trans-isomer reaction channels. When the enamine was in the s-cis-conformation, the transition states leading to the products all showed distortion of the s-cis-enamine framework, resulting in higher energies. In these transition states, other effects that contributed to the higher energy were seen, such as steric repulsion between the methyl group of the s-cis-enamine and benzaldehyde, and distortion of the aspartic acid substituent of the s-cis-enamine. In TS3-cR, the favorable interactions of the aspartic acid substituent with benzaldehyde, the hydrogen bond of the NH group with carbonyl were very similar to the one found for TS3-tR. However, to accommodate these interactions in an optimal way, the peptide backbone was rotated such that the methyl substitute of the enamine experienced steric repulsion from both the five-membered ring and the peptide bond. In the s-cis-enamine version of this transition state, it was possible to avoid this steric repulsion, but this came at the price of losing the interaction with the enamine proton, which resulted in a very similar energy. These effects led to energies that were considerably higher than those transition states obtained via the s-trans-enamine reaction channel. This higher energy was thus the source of the enantioselectivity.
For the final S-β-hydroxyketone product, two transition states were found to have energies both lying at 6.65 and 7.58 kcal mol−1 higher than TS3-tR. The lowest transition state leading to the S-β-hydroxyketone enantiomer of product TS3-tS had an energy of 6.65 kcal mol−1 higher than TS3-tR. However, changing the face of the benzaldehyde caused a ring–ring steric repulsion that raised the energy. Finally, in the highest transition state leading to the S-β-hydroxyketone product TS3-cS, two effects were responsible for raising the energy by 7.58 kcal mol−1 compared to TS3-tR. Previous theoretical studies showed that proton transfer from the amide group of the dipeptide to the forming intermediate was essential for charge stabilization and to facilitate C–C bond formation in the transition states.40
The relative energies of the transition states in the stereoselectivity-determining step leading to the two different stereoisomers were 0, 4.02, 6.65, and 7.58 kcal mol−1 for TS3-tR, TS3-cR, TS3-tS and TS3-cS, respectively. Theoretically, the R-product was found to be the major product, with an enantiomeric excess >99%, which translated approximately to an energy difference of 6.65 kcal mol−1 between the TS3-tR and TS3-tS transition states.41 This was due to the steric repulsion interaction between the carbonyl of the benzyl substituent and enamine terminus in the S-face attack. This also explained why the R-face transition state was preferred over the S-face for electrophilic attack.
In summary, the C–C bond and hydrogen bonding interactions made TS3-tR the optimal transition state, having a lower energy barrier than the other transition states leading to the final product. Taken together, the features of the steric repulsion interactions in other transition states discussed above jointly made TS3-tR the most stable transition state. The stabilization of the transition states provided by hydrogen bonding interactions was likely one of the main reasons that enable the dipeptide to catalyze the intermolecular aldol reaction with high enantioselectivity.
As discussed above, the main source of stereoselectivity was found to be the steric repulsion interactions of the enamines with benzaldehyde. The origin of the enantioselectivity arose from the distortion of the attack face of benzaldehyde and the distortion of the enamine backbone in the transition state geometries. B3LYP/6-31G(d,p) calculations matched the general experimental trends and provided useful insights into the origins of the variations in stereoselectivity.
 | ||
| Fig. 9 Structural models of final products. | ||
3.3 The energy variation in reaction path
It should be worth noting that there were two possible stereoselective channels including the form of the cis- and trans-isomerization in the aldol reaction. The energy variations via the O-path including the s-cis- and s-trans-enamine channels are shown in Fig. 10 and 11, respectively. The energies of acetone and dipeptide were set as 0.00 kcal mol−1 as a reference in the energy profile. According to the energy variation in the reaction path, the energy barrier of the first step was the maximum (44.33 kcal mol−1), so the first step was the rate-determining step. In the second step, there were three possible channels (C-path, N-path, O-path) and the O-path was the preferential channel in the aldol reaction from previous analysis. The corresponding former energy barrier of imine forming (dehydration) was 13.36 kcal mol−1, and the next energy barriers of proton transfer were 1.86 and 1.69 kcal mol−1 for the s-cis- and s-trans-enamines respectively. The energy barriers of proton transfer of the imine in the O-path were 1.86 (s-cis-enamine) and 1.69 (s-trans-enamine) kcal mol−1 (Table 1). It was small enough to be neglected in the course of the energy variation in Fig. 10 and 11. Subsequently, the activation energy barrier in the TS3-tR path was 6.65 kcal mol−1 lower than that in the TS3-tS path and the energy barrier in the TS3-cR path was 3.56 kcal mol−1 lower than that in the TS3-cS path of two channels which demonstrated that the R-product was the major product. Thus, the high ee value (>99%) could be explained by the larger energy difference (6.65 kcal mol−1) between the TS3-tS and TS3-tR transition states. Therefore, the O-path was thermodynamically more favorable, supporting the preference of reaction through the conformation of the s-trans-enamine path leading to the R-product.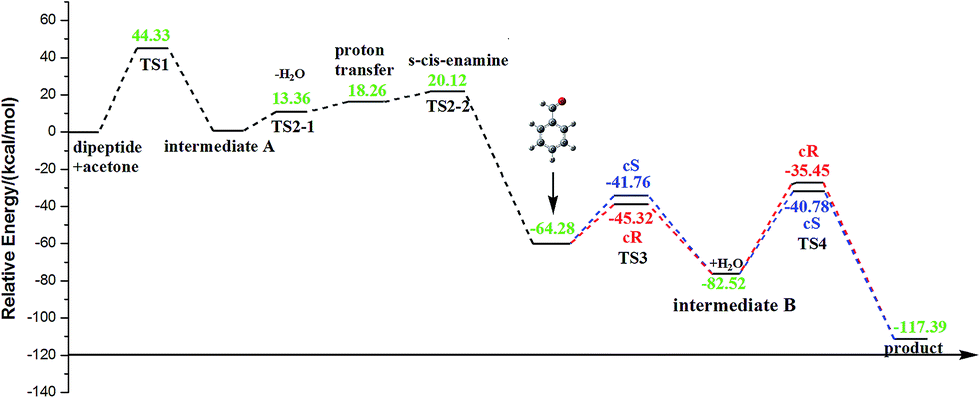 | ||
| Fig. 10 The energy variation via s-cis-enamine channel in O-path. | ||
 | ||
| Fig. 11 The energy variation via s-trans-enamine channel in O-path. | ||
| ΔE(B3LYP)/(kcal mol−1) | ΔE(CAM-B3LYP)/(kcal mol−1) | ΔE(M06-2X)/(kcal mol−1) | |
|---|---|---|---|
| TS1 | 44.33 | 45.23 | 41.65 |
| TS2-1 | 13.36 | 16.09 | 17.20 |
| TS2-2 (cis-) | 1.86 | 3.11 | 4.07 |
| TS2-2 (trans-) | 1.69 | 2.53 | 2.60 |
| TS3-tR | 14.94 | 14.94 | 11.83 |
| TS3-cR | 18.96 | 18.66 | 15.24 |
| TS3-tS | 21.59 | 21.90 | 21.37 |
| TS3-cS | 22.52 | 22.83 | 19.86 |
| TS4-tR | 42.74 | 39.97 | 38.46 |
| TS4-cR | 42.07 | 39.04 | 36.38 |
| TS4-tS | 42.58 | 39.54 | 36.90 |
| TS4-cS | 41.74 | 38.72 | 34.66 |
More specifically, we focused our attention on the nucleophilic attack of the chiral enamine at the carbonyl group of benzaldehyde, since the stereochemistry of the product was controlled in the step. In the equilibrium between the benzaldehyde and the enamine, an intermediate B was formed. However, to achieve this N–H⋯O hydrogen transfer, there was a significant loss in conjugation between the nitrogen lone pair and the CC double bond. The mechanism involved the attack of an enamine intermediate accompanied by proton transfer from the amide moiety to the developing benzaldehyde.
Furthermore, two other methods (CAM-B3LYP and M06-2X) were used to calculate the energy variations in the reaction path at the same basis set level of 6-31G(d,p). CAM-B3LYP combined the hybrid qualities of B3LYP and the long-range correction, while performed well for the charge transfer calculation in the dipeptide model.42 M06-2X, a hybrid meta-GGA functional, showed excellent performance for main group thermochemistry and noncovalent interactions, predicting accurate valence energies. A benchmark paper by Truhlar showed the good performances of the M06-2X functional for thermodynamic and kinetic properties,43 which agreed with their evaluation showing that M06-2X outperformed the B3LYP functional.44–46 M06-2X was chosen for its increased accuracy with main group atom energies and reaction kinetics, as well as for its ability to describe these medium-range electron correlations and Van der Waals interactions much more effectively than B3LYP.47 Previous studies indicated that the M06-2X functional could properly describe the thermodynamic stability within a series of transition states, hence, this was the functional chosen for this paper.48–51
The geometries of the dipeptide, reactants, transition states, intermediates and products were calculated with the M06-2X and CAM-B3LYP methods via the O-path. The energy barriers in the O-path calculated at the CAM-B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) levels in the gas phase are listed in Table 1. The M06-2X method aimed to improve the thermodynamic data for the aldol reaction using a functional that was expected to overcome the known deficiencies of the B3LYP functional when considering medium-range electron correlation effects. Past research showed that CAM-B3LYP and M06-2X calculations were in closer agreement with the available experimental data than the B3LYP calculations.52 We considered that due to the prevalence of hydrogen in chemical systems, the good reproduction of the total atomic energies, hydrogen in particular, was more important than the slight loss in quality of the atomization and ionization energies.
As shown in Table 1, the energy variation tendency was practically analogical using such methods. For the energy variation in reaction path using two correction functionals, the energy barrier of the first step was also the maximum (45.23 and 41.65 kcal mol−1, respectively), so the first step was the rate-determining step. In the second step, the O-path was the preferential channel in the aldol reaction from previous analysis. The energy barrier of dehydration of intermediate A calculated by CAM-B3LYP was increased, while it decreased with the M06-2X method. The disparities of the energy barriers to form two enamines were enlarged using two correction functionals which proved that the s-trans-enamine was energetically favorable. Additionally, the activation energy of TS3-tR was lower than the other three transition states which demonstrated that the R-product was the major product calculated by three different functionals. Accordingly, the conformation of the s-trans-enamine was thermodynamically more favorable, supporting the preference of the reaction leading to the R-product.
3.4 Solvation effects
According to the mechanism, this reaction occurred under solvent-free conditions at room temperature. Generally, the aldol reaction is operated in a solvent, and therefore, consideration of the solvent effects is crucial. In order to investigate the possible solvent effects, DMSO (εDMSO = 46.8) and H2O (εH2O = 78.4) were chosen as solvents using the PCM model in our calculations.Table 2 displays the energy barriers in the O-path calculated at the B3LYP/6-31G(d,p) level in the gas phase and in H2O and DMSO using the PCM model. Inclusion of the solvent DMSO and H2O reduced the activation energy barriers for stereoselective intermediate formation significantly, and yielded a greater exothermic reaction and stable R-product. However, the activation energy barriers of two conformations of enamines via a proton transfer process were increased. In order to illustrate this phenomenon, the proton transfer process of forming the s-trans-enamine in the gas phase and DMSO via the O-path is presented in Fig. 12 as an example.
| ΔE(gas)/(kcal mol−1) | ΔE(H2O)/(kcal mol−1) | ΔE(DMSO)/(kcal mol−1) | |
|---|---|---|---|
| TS1 | 44.33 | 37.61 | 40.60 |
| TS2-1 | 13.36 | 10.77 | 8.15 |
| TS2-2 (cis-) | 1.86 | 5.16 | 7.84 |
| TS2-2 (trans-) | 1.69 | 5.93 | 9.19 |
| TS3-tR | 14.94 | 11.98 | 12.04 |
| TS3-cR | 18.96 | 15.32 | 15.55 |
| TS3-tS | 21.59 | 15.14 | 17.14 |
| TS3-cS | 22.52 | 15.43 | 17.06 |
| TS4-tR | 42.74 | 40.46 | 5.07 |
| TS4-cR | 42.07 | 40.00 | 11.42 |
| TS4-tS | 42.58 | 40.10 | 5.63 |
| TS4-cS | 41.74 | 41.46 | 8.86 |
 | ||
| Fig. 12 Proton transfer to form s-trans-enamine in gas phase and DMSO via O-path. | ||
As shown in Fig. 12, the proton transferred to the O(5) atom forming the s-trans-enamine, in the meantime, the hydrogen bond between the dipeptide amide N(4) and carbonyl existed. In the gas phase, the hydrogen bond (N–H⋯O) distance was 1.992 Å, while it was 2.416 Å in DMSO which almost dissociated. The higher activation energy barrier was due to the hindrance of the hydrogen bond in solvent. The close similarity of the barriers for the dipeptide-catalyzed aldol reaction in H2O and DMSO in contrast to the catalytic effect highlights once more the importance of explicit solute–solvent interactions.
The geometries of transition state TS3-tR in the gas phase and DMSO are given in Fig. 13. In DMSO, the activation energy (12.04 kcal mol−1) needed for the formation of transition state TS3-tR was smaller than that (14.94 kcal mol−1) calculated in the gas phase. The effect of the solvent (DMSO) was predicted to further favor R-product formation, and thus a stereoselective reaction, even more.
 | ||
| Fig. 13 Structural model of TS3-tR in gas phase and DMSO. | ||
Overall, whether in the gas phase or in DMSO and H2O, there was obviously excellent agreement between the predicted and observed stereoselectivities for the dipeptide-catalyzed intermolecular aldol reaction. The relative energies of TS4 were decreased acutely in DMSO which demonstrated that DMSO was an advantageous solvent for the dipeptide catalyzed asymmetric aldol reaction experimentally.
3.5 Substituent effects of aromatic aldehydes
As shown in Table 3, the energy variations in the reaction path of the aldol reaction between acetone and three aromatic aldehydes calculated at the B3LYP/6-31G(d,p) level in the gas phase. All calculations of frequency with these transition state geometries yielded one and only one imaginary frequency and all real local minima. The data in Table 3 shows that the change of the substituent from a H atom to methyl-(electron donating group) and nitro-(electron withdrawing group) only had slight influences on the formation of the transition states. Similar to benzaldehyde, the lowest energy transition state was predicted to be TS3-tR in the stereoselectivity determining step, corresponding to the generation of the final product, R-β-hydroxyketone, which was in agreement with the experimental observations. The formation of a dominating R-product was nearly unaffected by the change of the substituent of the aldehydes due to the faint inductive effects of the p-position in the benzene ring and hence similar values for enantiomeric access (>99%) were observed for the aldol reactions. These results suggested that the introduction of a substituent at the p-position of benzaldehyde effectively provided the desired stereoselectivity for the asymmetric intermolecular aldol reaction.| ΔE(benzaldehyde)/(kcal mol−1) | ΔE(p-methyl benzaldehyde)/(kcal mol−1) | ΔE(p-nitrobenzaldehyde)/(kcal mol−1) | |
|---|---|---|---|
| TS3-tR | 14.94 | 15.59 | 12.38 |
| TS3-cR | 18.96 | 19.42 | 16.66 |
| TS3-tS | 21.59 | 22.14 | 18.80 |
| TS3-cS | 22.52 | 23.08 | 19.12 |
| TS4-tR | 42.74 | 42.32 | 42.58 |
| TS4-cR | 42.07 | 42.16 | 41.59 |
| TS4-tS | 42.58 | 42.56 | 42.61 |
| TS4-cS | 41.74 | 41.61 | 42.23 |
It was useful to confirm the HOMO and the LUMO of the transition states in the stereoselectivity-determined step. The relative ordering of the occupied and virtual orbitals provided a reasonable qualitative indication of the excitation properties. The HOMO and LUMO energies of these transition states in the stereoselectivity determining step are listed in Table 4. Table 4 displays that the order of transition states for the HOMO orbital energies using three aromatic aldehydes was E(p-methyl benzaldehyde) > E(benzaldehyde) > E(p-nitrobenzaldehyde), indicating that the introduction of methyl- in the p-position of benzaldehyde enhanced the HOMO orbital energy, while the nitro-substituent reduced the HOMO orbital energy. The faint inductive effects in the benzene ring due to the p-position substituent meant that a methyl-group could enhance the electronic density as an electron donating group, on the contrary, a nitro-group could reduce the electronic density as an electron withdrawing group. The results proved that the different substituents in the p-position of the benzene ring initiated the inductive effects; however the p-position was relatively distant so had only a slight influence on the energy barriers and no influence on the stereoselectivity of the major product. From the comparison of energy barriers, the value of the same transition state of the methyl-substituent increased and that of the nitro-substituent decreased as a result of the introduction of a substituent in the p-position of benzaldehyde. The electronic density contours of the frontier orbitals (HOMO and LUMO orbitals) of the transition states in the stereoselectivity determining step are plotted in the ESI.†
| HOMO (eV) | LUMO (eV) | Gap (eV) | ||
|---|---|---|---|---|
| Benzaldehyde | TS3-tR | −0.229 | −0.076 | 0.153 |
| TS3-cR | −0.235 | −0.075 | 0.160 | |
| TS3-tS | −0.228 | −0.080 | 0.148 | |
| TS3-cS | −0.223 | −0.079 | 0.144 | |
| p-Methyl benzaldehyde | TS3-tR | −0.225 | −0.074 | 0.151 |
| TS3-cR | −0.231 | −0.074 | 0.157 | |
| TS3-tS | −0.225 | −0.078 | 0.147 | |
| TS3-cS | −0.219 | −0.077 | 0.142 | |
| p-Nitro benzaldehyde | TS3-tR | −0.241 | −0.096 | 0.145 |
| TS3-cR | −0.246 | −0.098 | 0.148 | |
| TS3-tS | −0.238 | −0.098 | 0.140 | |
| TS3-cS | −0.233 | −0.096 | 0.137 |
4. Conclusions
The mechanism of the dipeptide-catalyzed intermolecular aldol reaction was investigated using different DFT methods. There were four steps in the reaction path, the first step was the rate-determining step due to it having the largest energy barrier of 44.33 kcal mol−1, but the third step decided the stereoselectivity of the product because the steric repulsion of functional groups governed the stereoselectivity of the products severely. The effects of leading the R-product through the s-trans-isomer path were the steric repulsion between the aspartic acid substitute of the enamine and the aldehyde; and the steric repulsion between the attack face of the aldehyde and the enamine. It was found that the dipeptide-catalyzed aldol reaction via an s-trans-enamine was more energetically favorable to obtain the R-product (with an ee value > 99%). Additionally, the computed results provided a reference for experiments that full mixing of the catalyst and reactants or operation at elevated temperatures was essential and DMSO or H2O as the solvent could decrease the energy barriers in the reaction path. We displayed that the CAM-B3LYP and M06-2X density functionals performed as well as and better than the B3LYP functional for charge transfer transition states with steric effects but had better performance for bond energies, noncovalent interactions, and chemical reaction barrier heights for representative systems. The introduction of a substituent at the p-position of benzaldehyde had a slight influence on the energy barriers and no influence on stereoselectivity of the major product for the asymmetric intermolecular aldol reaction. Owing to few researches that were concerned with the detailed variations of the entire path, we concluded that the dipeptide should be preferred for studies requiring the exploration of potential green syntheses as well as nontoxic catalysts, provided that the dipeptide of this sort of protein are included.Acknowledgements
This project was supported by the National Natural Science Foundation of China (21173019).Notes and references
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 31, 325–335 CrossRef.
- Z. Tang, F. Jiang, L. T. Yu, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, J. Am. Chem. Soc., 2003, 125, 5262–5263 CrossRef CAS PubMed.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed.
- D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS PubMed.
- S. Gandhi and V. K. Singh, J. Org. Chem., 2008, 73, 9411–9416 CrossRef CAS PubMed.
- X. H. Liu, L. L. Lin and X. M. Feng, Chem. Commun., 2009, 41, 6145–6158 RSC.
- C. D. Maycock and M. R. Ventura, Tetrahedron: Asymmetry, 2012, 23, 1262–1271 CrossRef CAS.
- A. K. Pandey, D. Naduthambi, K. M. Thomas and N. J. Zondlo, J. Am. Chem. Soc., 2013, 135, 4333–4363 CrossRef CAS PubMed.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1375 CrossRef CAS.
- G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418–7421 CrossRef CAS PubMed.
- H. J. Martin and B. List, Synlett, 2003, 12, 1901–1902 Search PubMed.
- F. Kolundzic, M. N. Noshi, M. Tjandra, M. Movassaghi and S. J. Miller, J. Am. Chem. Soc., 2011, 133, 9104–9111 CrossRef CAS PubMed.
- S. Bayat, B. A. Tejo, A. B. Salleh, E. Adbmalek, Y. M. Normi and M. B. A. Rahman, Chirality, 2013, 25, 726–734 CrossRef CAS PubMed.
- T. Zhuo, Z. H. Yang, L. F. Cun, L. Z. Gong, A. Q. Mi and Y. Z. Jiang, Org. Lett., 2004, 6, 2285–2287 CrossRef PubMed.
- W. Zou, I. Ibrahem, P. Dziedzic, H. Sundén and A. Córdova, Chem. Commun., 2005, 39, 4946–4948 RSC.
- E. Machuca and E. Juaristi, Tetrahedron Lett., 2015, 56, 1144–1148 CrossRef CAS.
- P. Dziedzic, W. Zou, J. Hafren and A. Córdova, Org. Biomol. Chem., 2006, 4, 38–40 CAS.
- A. Bassan, W. Zou, E. Reyes, F. Himo and A. Córdova, Angew. Chem., Int. Ed., 2005, 44, 7028–7032 CrossRef CAS PubMed.
- A. Córdova, W. Zou, I. Ibrahem, E. Reyes, M. Engqvist and W. W. Liao, Chem. Commun., 2005, 36, 3586–3588 RSC.
- A. Heine, G. Desantis, J. G. Luz, M. Mitchell, C.-H. Wong and I. A. Wilson, Science, 2001, 294, 369–374 CrossRef CAS PubMed.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., 2000, 112, 1406–1430 CrossRef.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386–7387 CrossRef CAS.
- M. Amedjkouh, Tetrahedron: Asymmetry, 2005, 16, 1411–1414 CrossRef CAS.
- M. Limbach, Tetrahedron Lett., 2006, 47, 3843–3847 CrossRef CAS.
- S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 12911–12912 CrossRef CAS PubMed.
- L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16–17 CrossRef CAS PubMed.
- B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827–833 CrossRef CAS PubMed.
- C. Allemann, J. M. Um and K. N. Houk, J. Mol. Catal. A: Chem., 2010, 324, 31–38 CrossRef CAS PubMed.
- F. R. Clemente and K. N. Houk, J. Am. Chem. Soc., 2005, 127, 11294–11302 CrossRef CAS PubMed.
- Y. K. Zhang, J. Zhu, N. Yu and H. Yu, Chin. J. Chem., 2015, 33, 169–174 Search PubMed.
- K. Liu and G. Zhang, Tetrahedron Lett., 2015, 56, 243–246 CrossRef CAS.
- S. S. V. Ramasastry, H. Zhang, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2007, 129, 288–289 CrossRef CAS PubMed.
- S. Pizzarello and A. L. Weber, Science, 2004, 303, 1151 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gau-ssian 09 Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Fu, H. Li, F. Tian, S. Yuan, H. Si and Y. Duan, Tetrahedron: Asymmetry, 2008, 19, 1288–1296 CrossRef CAS.
- B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839–5842 CrossRef CAS PubMed.
- G. Yang, Z. Yang, L. Zhou, R. Zhu and C. Liu, J. Mol. Catal. A: Chem., 2010, 316, 112–117 CrossRef CAS.
- P. Hammar, A. Córdova and F. Himo, Tetrahedron: Asymmetry, 2008, 19, 1617–1621 CrossRef CAS.
- O. B. Gadzhiev, L. A. G. D. L. Rosa, F. J. M. Bustamante, C. A. D. Parrodi, H. H. Abdallah, A. I. Petrov and T. Scior, J. Phys. Org. Chem., 2012, 25, 971–978 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. D. Bose and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef PubMed.
- S. Rayne and K. Forest, THEOCHEM, 2010, 948, 102–107 CrossRef CAS.
- D. H. Ess, S. Liu and F. D. Proft, J. Phys. Chem. A, 2010, 114, 12952–12957 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Org. Lett., 2006, 8, 5753–5755 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- R. Huenerbein, B. Schirmer, J. Moellmann and S. Grimme, Phys. Chem. Chem. Phys., 2010, 12, 6940–6948 RSC.
- R. Li, J. Zheng and D. G. Truhlar, Phys. Chem. Chem. Phys., 2010, 12, 12697–12701 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26808g |
| This journal is © The Royal Society of Chemistry 2016 |