
Selective separation and identification of metabolite groups of Polygonum cuspidatum extract in rat plasma using dispersion solid-phase extraction by magnetic molecularly imprinted polymers coupled with LC/Q-TOF-MS†
Abstract
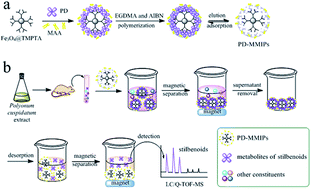
In this work, magnetic molecularly imprinted polymers (MMIPs) were successfully prepared for specific recognition and selective enrichment of metabolite groups of Polygonum cuspidatum extract in rat plasma. Two kinds of MMIPs were obtained using polydatin (PD) and emodin-8-O-β-D-glucoside (EG) as template molecules, respectively, methacrylic acid as functional monomer, ethylene glycol dimethacrylate as cross-linker and 2,2′-azobisisobutyronitrile as initiator. The membrane thickness of both MMIPs was approximately 10 nm. With the dendritic-grafting modification, magnetic nanoparticles (MNPs) possessed the outstretched branched structure, orderly open cavities and abundant functional groups with a good specific surface area (47.99 m2 g−1). And both MMIPs obtained high adsorption capacity (221 μmol g−1 for PD and 190 μmol g−1 for EG) and fast kinetics. Dispersion solid-phase extraction using MMIPs (MMIPs-DSPE) was applied to selectively separate and enrich the target metabolite groups of stilbenoids and anthraquinones, respectively. Furthermore, three classical pretreatment methods including protein precipitation (PP), liquid–liquid extraction (LLE) and solid phase extraction (SPE) were compared with MMIPs-DSPE in terms of matrix effect, efficiency of extraction and capacity using LC/Q-TOF-MS. The result showed that MMIPs obtained the lowest matrix effect from 1.52 to 8.96% and the most constituents of target metabolite groups with 17 stilbenoids and 19 anthraquinones, respectively. The proposed analysis platform has been proven to be selective, sensitive and simple for the enrichment of target metabolite groups, and has great significance for discovering minor bioactive constituents and elucidating integrative mechanism of traditional Chinese medicines.
 Please wait while we load your content...
Please wait while we load your content...

