
Synthesis, photophysics, ion detection and DFT investigation of novel cyano-substituted red-light chromophores with a triphenylamine donor†
Abstract
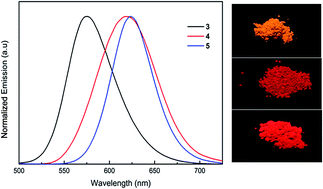
A series of novel triphenylamine-based red-light chromophores with multiple electron-withdrawing cyano substituents were synthesized by the Knoevenagel condensation reaction and characterized in detail. Compounds 3–5 showed bright green-yellow emission in dichloromethane solution and red-light emission in the solid state, respectively. The interesting solvatochromic behavior in different polar solvents was observed, varying from positive solvatochromism for compound 3 to negative solvatochromism for compound 5. In addition, the notable optical response of metal ions in DMF solution for the cyano-substituted chromophores was investigated. Especially, with the addition of Hg2+ ions, the blue shift in the absorption spectra and the decrease in the emission spectra suggested that the new metal complexes possibly formed between the cyano substituents and metal ions or metal ion-induced optical quenching happened. Density functional theory calculations were used to further understand the effect of the cyano substituents on the photoelectron properties of the donor–acceptor molecules.
 Please wait while we load your content...
Please wait while we load your content...

