DOI:
10.1039/C5RA26674B
(Paper)
RSC Adv., 2016,
6, 16520-16532
Atorvastatin calcium loaded PCL nanoparticles: development, optimization, in vitro and in vivo assessments†
Received
14th December 2015
, Accepted 1st February 2016
First published on 2nd February 2016
Abstract
The aim of the present study was to prepare atorvastatin calcium (ATR) loaded poly(ε-caprolactone) nanoparticles (ALPNs) to enhance the oral bioavailability, efficacy and safety profile of drug. ALPNs were prepared by a nanoprecipitation technique while formulation and process parameters were optimized using a central composite factorial design. The optimized ALPNs were investigated through in vitro (solid state characterization, morphological, drug release study and stability study) analysis and in vivo (pharmacokinetic, efficacy and safety study) behaviour in rats. The optimized ALPNs having 197 ± 5 nm particle size, 0.213 ± 0.012 polydispersity index and 75.6 ± 3.2% entrapment efficiency, did not exhibit any physicochemical interaction of the drug with the carrier. The X-ray diffraction, differential scanning calorimetry and electron diffraction pattern has substantiated the amorphous character of ATR encapsulated in nanoparticles. The smooth and homogeneous spherical shape of the nanoparticles was evidenced in morphological analyses. The in vitro drug release profile of ALPNs showed a 96 h sustained release and the pharmacokinetic profile in rats exhibited significant enhancement in bioavailability, Cmax and mean resident time of the drug. ALPNs exhibited similar efficacy (plasma lipid profile and glucose level) and markedly improved biochemical safety profiles (creatinine, blood urea nitrogen, creatinine kinase, lactate dehydrogenase and aspartate amino transferase) of rat plasma at a 50% reduced dose compared to orally administered ATR.
1. Introduction
Biodegradable polymeric nanoparticles (PNs) have drawn considerable attention from scientists, owing to their promising advantages over other carrier systems.1 A wide variety of therapeutic agents have been encapsulated in PNs and proved to be a competent novel delivery system toward improved stability, a sustained release profile, oral bioavailability, efficacy along with reduced toxicity of the therapeutic agent.2–4 PNs can be easily prepared by simple, robust and non-invasive versatile techniques with smaller uniform particle sizes and good storage stability. Surface property of PNs can be tailored in order to protect them from opsonisation and target the drug loaded PNs to particular organs or tissues.5–7 PNs not only protect the sensitive drug from hydrolytic and enzymatic degradation in gastro-intestinal tract (GIT) but also follow the lymphatic pathway via M-cells of Payer's patches in GIT to bypass the first-pass metabolism.8 PNs can improve safety profile of drug by sustaining the drug release as well as reducing the dose size or dosing frequency on the basis of enhanced bioavailability.
Atorvastatin calcium (ATR) is a widely prescribed statin drug for controlling of hyperlipidemia and atherosclerosis on account of its greater potency, efficacy and other therapeutic properties. Hyperlipidemia, a metabolic syndrome caused by elevated levels of plasma lipids, is a crucial risk factor for atherosclerosis.9 ATR not only inhibits 3-hydroxy-3-methylgluteryl coenzyme A reductase (a rate limiting enzyme in cholesterol biosynthesis) but also arrests vascular smooth muscle cell proliferation and platelet hyperfunction for limiting the atherosclerosis and superadded thrombosis, respectively.10,11 It has also been indicated for antioxidant, anti-inflammatory and anticancer activity.12–14 In spite of being a megahit drug, its oral bioavailability is limited to only 12% as a result of low solubility and high presystemic elimination by first pass metabolism as well as gut wall efflux pump.15,16 Cytochrome P-450 3A4 enzyme extensively found in gut wall and hepatic region is responsible for metabolism of ATR. Patients suffering from hyperlipidemia and atherosclerosis require a longer ATR treatment regime with other drug combination. However, chronic use of ATR leads to skeleton muscle toxicity from mild myalgia to rhabdomyolysis, which compels the patients to remain non-adherent with prescribed pharmacotherapy.17
ATR comes under class II drug as per biopharmaceutical classification system.18 Several formulation approaches have been attempted in past with an objective to improve ATR bioavailability and efficacy. Several research groups have reported self micro/nano emulsifying drug delivery systems of ATR with improved bioavailability.19–23 Solid dispersion of ATR with polyvinylpyrrolidone vinyl acetate was reported for better bioavailability due to enhanced supersaturation and dissolution.24 Amorphous nanoparticles of ATR with enhanced bioavailability were also reported.25,26 The reported chitosan conjugated ATR and its nanoparticles exhibited markedly improved bioavailability than the pure drug.27 Polymeric nanoparticles based on poly(lactic-co-glycolic acid) of ATR reported to sustain the drug release profile and to enhance oral bioavailability.28 Nevertheless, all the formulation strategies lack with sufficient safety and efficacy studies in order to successful translation in the clinical settings.
Poly(ε-caprolactone) (PCL) is a biocompatible and biodegradable United States food and drug administration approved synthetic polymer, which is widely used as drug delivery carriers and devices.29 PCL based PNs can provide a longer sustained drug release profile owing to its higher molecular weight and hydrophobicity. D-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) has been extensively used as a stabiliser on account of its additional antioxidant and bioavailability enhancing property. Surface absorbed TPGS molecules may impart hydrophilic characteristics due to PEG and provide prolong retention of nanoparticles in the systemic blood circulation.30 On extensive literature survey, no research regarding PCL based PNs of ATR using TPGS as stabiliser has been reported yet. Hence, ATR loaded PCL nanoparticles (ALPNs) with TPGS as stabiliser, have been hypothesized for enhancing ATR bioavailability, efficacy and safety profiles. Nowadays, quality by design has become an integral part of formulation and product development. Central composite design (CCD), a response surface methodology (RSM) model, has been extensively used as quality by design tool for optimization of nanoparticles' formulation and process parameters.28,31–33 In the present study, CCD has been employed to optimize the several critical variables of ALPNs. Further, optimized ALPNs have been extensively evaluated for numerous in vitro and in vivo characterizations in order to substantiate the hypothesis.
2. Materials and methods
2.1 Materials
Atorvastatin calcium and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) were obtained as a kind gift sample from Ipca Laboratories Limited, Dehradun and Antares Health Product Inc., Mumbai respectively. PCL (MW ∼ 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Sigma-Aldrich, USA. Disodium hydrogen orthophosphate AR, potassium dihydrogen orthophosphate AR, acetone HPLC grade, methanol HPLC grade and phosphoric acid HPLC grade were purchased from Merck Limited, Mumbai. All the diagnostic kits for lipid profile [plasma triglyceride (PTG), plasma total cholesterol (PTC) and high density lipoprotein-cholesterol (HDL-C)], glucose, creatinine kinase (CK), lactate dehydrogenase (LDH), blood urea nitrogen (BUN) and creatinine level were purchased from Span Diagnostic Ltd., Surat. All other chemicals and solvent were of analytical grade and procured from SD fine-chem Ltd., Mumbai.
000 Da) was purchased from Sigma-Aldrich, USA. Disodium hydrogen orthophosphate AR, potassium dihydrogen orthophosphate AR, acetone HPLC grade, methanol HPLC grade and phosphoric acid HPLC grade were purchased from Merck Limited, Mumbai. All the diagnostic kits for lipid profile [plasma triglyceride (PTG), plasma total cholesterol (PTC) and high density lipoprotein-cholesterol (HDL-C)], glucose, creatinine kinase (CK), lactate dehydrogenase (LDH), blood urea nitrogen (BUN) and creatinine level were purchased from Span Diagnostic Ltd., Surat. All other chemicals and solvent were of analytical grade and procured from SD fine-chem Ltd., Mumbai.
2.2 Formulation of ALPNs
Nanoprecipitation technique was employed for the preparation of ALPNs. Concisely, intended amount of PCL and ATR were dissolved in acetone and this organic phase was added at the rate of 2 ml min−1 with the help of syringe (needle size #23) to TPGS aqueous solution while keeping the aqueous solution under magnetic stirring. The mixture was kept continuously under magnetic stirring overnight to evaporate acetone. The resulting nanoparticle suspension was centrifuged at 16000 rpm for 30 min and the sediment nanoparticles were resuspended in 0.2% (w/v) TPGS aqueous solution. The prepared ALPNs were subjected to in vitro and in vivo evaluation. The suspension was lyophilized with 2% (w/v) mannitol by using freeze-drier (LABCONCO, USA). The effect of lyophilisation on ALPNs properties (PS, PDI and EE) were estimated by comparing these properties prior and post lyophilisation of ALPNs dispersion. Samples of ALPNs subjected for solid state characterization were lyophilized deliberately without using mannitol to avoid possible interference of mannitol thermogram and diffractogram with ATR thermogram and diffractogram.
2.3 Central composite design and characterization of ALPNs
Four factors (X1) polymer content, (X2) TPGS concentration, (X3) volume of acetone and (X4) stirring speed were selected as formulation variables on the basis of preliminary formulation study. These four factors with five coded levels (−2, −1, 0, +1 and +2) were selected as independent formulation variables whereas mean diameter hydrodynamic particle size (R1), mean entrapment efficiency (R2) and mean polydispersity index (R3) were selected as depended response variables, respectively. All the independent variables with their actual and coded values are shown in Table 1. CCD-RSM was applied for optimization of formulation variables using Design-Expert 7.0 (State-ease Inc., USA) software. 30 batches consisting of 16 factorial points (level ‘−1’ and ‘+1’), 8 axial points (level ‘−2’ and ‘+2’) and 6 replicated central points (level ‘0’) were developed to establish a mathematical relation which expressed each of response variables as a function of independent formulation variables as shown in eqn (1).| | |
R = Ao + A1X1 + A2X2 + A3X3 + A4X4 + A12X1X2 + A13X1X3 + A14X1X4 + A23X2X3 + A24X2X4 + A34X3X4 + A11X12 + A22X22 + A33X32 + A44X42 + A
| (1) |
where R represents the dependent response variables (R1, R2 and R3), Ao is an intercept, Ai are linear terms, Aij (i and j = 1, 2, 3 and 4, i < j) represents the interaction terms, Aii represents the quadratic terms and A is random error.
Table 1 List of selected formulation and response variables with coded and exact valuea
| Factors |
Levels |
| Independent formulation variables |
−2 |
−1 |
0 |
1 |
2 |
| PCL: poly(ε-caprolactone), TPGS: D-α-tocopheryl poly(ethylene glycol) 1000 succinate. |
| (A) PCL (mg) |
30 |
60 |
90 |
120 |
150 |
| (B) TPGS concentration (w/v%) |
0.2 |
0.4 |
0.6 |
0.8 |
1.0 |
| (C) Volume of acetone (ml) |
3 |
5 |
7 |
9 |
11 |
| (D) Stirring speed (rpm) |
500 |
750 |
1000 |
1250 |
1500 |
| Dependent response variables |
Constraints |
| R1 = mean diameter particle size (nm) |
Minimize |
| R2 = mean entrapment efficiency (%) |
Maximize |
| R3 = mean polydispersity index |
Minimize |
Correlation co-efficient (r2) indicates quality of mathematical relation whereas analysis of variance (p < 0.05) indicates the significance of the model as well as its various linear (Xi), interaction (Xij) and quadratic (Xi2) terms. Optimal values of independent formulation variables of optimized batch were obtained by setting desired constraint in optimization tool.
2.4 Particle size, polydispersity index, zeta potential and entrapment efficiency determination
Hydrodynamic mean diameter particle size (PS) and polydispersity index (PDI) analysis were carried out by photon correlation spectroscopy (PCS) using particle size analyser (Delsa Nano C, Beckman Coulter Counter, USA) equipped with software N4 plus. The measurements were performed at an angle of 165° at 25 °C with proper dilution with Milli-Q® water. The zeta potential was analysed by measuring electrophoretic light scattering using the same particle size analyser in zeta potential mode.
Entrapment efficiency of ALPNs suspension was analysed by indirect method as reported by Ravi et al.34 Five ml of ALPNs sample was centrifuged for 30 minutes in cooling centrifuge (Remi, India) at 15000 rpm at 4 °C. The supernatant was analysed for free dissolved drug (FDD) by UV-Visible spectrophotometer (Shimadzu 1700, Japan) at 247 nm wavelength.
EE of ALPNs was determined using following formula
| |
 | (2) |
where TAD: total amount of drug and FDD: free dissolved drug.
2.5 Solid state characterisation of ALPNs
Drug–polymer interaction study is a crucial step in formulation development. Each sample of pure ATR, physical mixture (PM) of ATR with PCL and TPGS, placebo-PCL nanoparticles and ALPNs were characterised by Fourier transform infra red (FTIR) spectroscopy, differential scanning calorimetry (DSC) and powder X-ray diffraction (PXRD) study. Physical mixture was comprised of ATR, PCL and TPGS in 1:3:0.04 proportions. Chemical interaction study of drug in ALPNs was analysed by using FT-IR spectrophotometer (Shimadzu 8400, Japan) in the range of 400–4000 cm−1. Samples were analysed by pressed pellet technique using KBr as diluent. DSC analyses of samples were recorded on TGA/DSC-1, Star-system (Mettler Toledo, Switzerland) at a heating rate of 10 °C min−1 with 50 ml min−1 nitrogen purging rate. Thermal behaviours of samples were investigated in the range of 30 °C to 250 °C. PXRD patterns were analysed to investigate crystalline/amorphous nature of samples by using portable X-ray diffractometer (Rigaku, Japan) with Cu rotating anode (Kα radiation; λ = 1.54 nm) generated at 18 kW.
2.6 Morphological analyses of ALPNs
Morphological studies were performed using transmission electron microscopy (TEM) and atomic force microscopy (AFM). A drop of freshly prepared ALPNs suspension on each of TEM copper grid and borosilicate glass cover slip were air dried at 25 °C to prepare a fine film on respective platform. TEM grid with prepared film was fixed into sample holder to observe the image in TEM instrument (PHILIPS TECHNAI-20G2, Japan) under low vacuum at high resolution accelerating voltage 200 kV. Observed TEM and electron diffraction (ED) pattern images were recorded. Cover glass slip with prepared film was mounted on sample holder and image was recorded in AFM (NT-MDT, Russia) using solver next software.
2.7 In vitro release study
Dialysis bag (Himedia labs, cutoff weight 12000–14000 Da) filled with ALPNs suspension equivalent to 2.5 mg of ATR was sealed and incubated in 100 ml of 50 mM phosphate buffer saline (PBS, pH 7.4) at 37 ± 0.5 °C under mild agitation. At each preset time interval (0, 0.5, 1, 2, 4, 8, 12, 16, 24, 32, 40, 48, 56, 64, 72, 84 and 96 h), 2 ml samples were withdrawn from released media and analysed spectrophotometrically at 247 nm. After each sampling, 2 ml of fresh PBS was replenished in the release media to maintain sink condition. Similar study was performed for the pure ATR suspension in 0.25% sodium carboxy methyl cellulose. Release data were fitted into zero order, first order, Higuchi, Korsmeyer–Peppas (KP) and Hixson–Crowell release kinetic models to investigate the release mechanism of drug from the ALPNs.35
2.8 Stability study
2.8.1 Stability study in simulated biological fluid. ALPNs stability was assessed in different biological media [simulated gastric fluid (SGF) and simulated intestinal fluid (SIF)] in order to determine the effect of different gastrointestinal pH and enzymatic condition on their characteristic properties (PS, EE and PDI). SGF was composed of 2 g of sodium chloride, 3.2 g of pepsin, 7 ml of concentrated hydrochloric acid and water sufficient to make 1000 ml; whereas SIF contained 6.8 g of monobasic potassium phosphate, 77 ml of 0.2 N sodium hydroxide, 10 g of pancreatin and water sufficient to make 1000 ml as per USP, 2007. Briefly, 2 ml of reconstituted lyophilized ALPNs was incubated in 10 ml of different simulated fluids at 37 °C for specified time interval.36 After incubation period, ALPNs were analyzed for PS, PDI and EE to assess their stability in simulated biological fluids.
2.8.2 Storage stability study. The stability of ALPNs was evaluated over a period of six months at accelerated condition (40 ± 2 °C/75 ± 5% RH), normal condition (25 ± 2 °C/60 ± 5% RH) and refrigerated condition (4 ± 1 °C) as per ICH guideline. Freshly prepared lyophilized ALPNs were sealed in amber colour glass vials and placed in stability chamber at above mentioned conditions. Samples were withdrawn at different time points (0, 1, 2, 3 and 6 month) and analyzed for its properties (PS, EE and PDI).
2.9 In vivo study
2.9.1 Animals. Male Charles Foster rats weighing (180–220 g) were procured from central animal house of Institute of Medical Sciences, Banaras Hindu University, Varanasi (India). Each group of rats (n = 6) were domiciled in propylene cage at an ambient temperature of 25 ± 1 °C, 45–55% relative humidity with a 12 h light/12 h dark cycle fed with normal rat diet and water ad libitum. All study protocols were approved by Central Animal Ethical Committee of Banaras Hindu University (Dean/13-14/CAEC/321).
2.9.2 Pharmacokinetic study. ATR drug suspension (DS) and ALPNs group of overnight fasted rats (n = 6) were selected for pharmacokinetic study. Pure drug and ALPNs suspensions equivalent to 20 mg kg−1 of ATR were administered orally to each rat of respective group. At predetermined time interval (0.5, 1, 2, 4, 8, 16 and 24 h) after dosing, blood samples (0.3 ml) were collected from orbital sinus of each rat into a heparinised micro-centrifuge tube and plasma were separated by cooling centrifugation at 4000 rpm for 5 min at 4 °C. ATR present in plasma samples were analysed as mentioned in reported RP-HPLC method.28 Pharmacokinetic parameters were analysed by non-compartmental analysis using Kinetica 5.0 pharmacokinetic software (PK-PD analysis, Thermofischer) and statistically analysed by student unpaired t test using GraphPad Prism 5.03 (GraphPad Software, USA).
2.9.3 Efficacy and safety study. Normal diet control (NDC), high fat-diet control (HFC), high fat-diet placebo-PCL-nanoparticles treated (HFPPT), high fat-diet ATR suspension treated (HFAST) and high fat-diet ALPNs treated (HFAPT) groups of rats (n = 6 per group) were selected for eight week study divided into three parts [(1st–4th week) hyperlipidemia induction period, (4th–6th week) treatment period and (6th–8th week) washout period]. All rats except NDC group were fed with high fat diet consisting of 68% normal rat feed, 10% lard, 10% egg-yolk powder, 10% sugar and 2% cholesterol. Rats encaged in NDC group were fed normal rat pellet diet. After 4 weeks, all groups were treated accordingly as mentioned in Table 2 during fourth to sixth week. Blood samples were withdrawn from each tested rat at the end of 2nd, 4th, 6th, 7th and 8th week and analysed for various biochemical parameters (PTC, PTG, HDL-C, CK, BUN, LDH, AST and creatinine) using commercially available span diagnostic kits. Very low density lipoprotein (VLDL-C) and low density lipoprotein (LDL-C) were calculated by using Friedewald formula. The atherosclerosis index (AI) was calculated by formula expressed in eqn (3).37| |
 | (3) |
Table 2 Treatment regimen during 4th–6th week to different groupa
| Group |
Diet during 8 week |
Treatment during 4th–6th week (mg per kg per day) |
| NDC: normal diet controlled; HFC: high fat-diet controlled; HFPPT: high fat-diet placebo-PCL-nanoparticles treated; HFAST: high fat-diet atorvastatin calcium suspension treated; HFAPT, high fat-diet ALPNs treated. |
| NDC |
Normal rat diet |
— |
| HFC |
High fat diet |
— |
| HFPPT |
High fat diet |
Placebo-PCL-nanoparticles equivalent of ALPNs weight treated to HFAPT |
| HFAST |
High fat diet |
3 mg per kg per day of ATR |
| HFAPT |
High fat diet |
Equivalent weight of ALPNs containing 1.5 mg of ATR |
Experimental animals were sacrificed and isolated liver tissues were examined for histology.
2.10 Statistical analysis
All the statistical comparison among groups were performed by using nonparametric unpaired Student's t test (pharmacokinetic parameters) and Bonferroni post test two way ANOVA (safety and efficacy parameters) with the use of GraphPad Prism 5.03 (GraphPad Software, USA) software. Difference with a value of p < 0.05 was considered to be statistically significant.
3. Results and discussion
3.1 CCD and characterization of ALPNs
30 batches of ALPNs (Table 3) were prepared by employing CCD RSM. Results indicated that all selected independent variables significantly determined PS, EE and PDI of ALPNs. The adjusted mathematical expression with r2 value of response variables as a function of independent variables are as follow| | |
Mean diameter hydrodynamic particle size (R1) = +201.7 + 33.7 × X1 − 16.1 × X2 − 15.2 × X3 − 16.7 × X4 + 4.7 × X1 × X2 − 4.5 × X1 × X4 + 13.6 × X12 + 6.7 × X32 (r2 = 0.98)
| (4) |
| | |
Mean entrapment efficiency (R2) = +75.0 + 9.4 × X1 − 2.9 × X2 − 3.7 × X3 − 2.5 × X4 + 2.9 × X1 × X3 + 1.8 × X12 (r2 = 0.92)
| (5) |
| | |
Mean PDI (R3) = +0.230 + 0.039 × X1 − 0.028 × X2 − 0.017 × X3 − 0.011 × X4 − 0.009 × X3 × X4 + 0.022 × X12 (r2 = 0.99)
| (6) |
Table 3 ALPNs batches designed by central composite designa
| Batch No. |
Independent formulation variables with coded value |
Dependent response variables |
| X1 |
X2 |
X3 |
X4 |
R1 (nm) |
R2 (%) |
R3 |
| X1: weight of PCL, X2: TPGS concentration, X3: volume of acetone, X4: stirring speed, R1: mean diameter particle size, R2: mean entrapment efficiency, and R3: mean polydispersity index. |
| 1 |
1 |
1 |
−1 |
1 |
235 |
82.9 |
0.274 |
| 2 |
0 |
0 |
0 |
−2 |
241 |
84.3 |
0.249 |
| 3 |
1 |
1 |
1 |
1 |
219 |
91.2 |
0.218 |
| 4 |
0 |
0 |
0 |
2 |
167 |
72.5 |
0.207 |
| 5 |
−1 |
−1 |
−1 |
1 |
211 |
75.9 |
0.252 |
| 6 |
0 |
0 |
0 |
0 |
192 |
78.6 |
0.225 |
| 7 |
−2 |
0 |
0 |
0 |
189 |
65.7 |
0.236 |
| 8 |
0 |
0 |
0 |
0 |
196 |
76.3 |
0.226 |
| 9 |
−1 |
1 |
−1 |
1 |
171 |
67.3 |
0.194 |
| 10 |
−1 |
−1 |
1 |
−1 |
206 |
69.2 |
0.24 |
| 11 |
2 |
0 |
0 |
0 |
324 |
95.3 |
0.396 |
| 12 |
1 |
−1 |
−1 |
−1 |
306 |
95.7 |
0.336 |
| 13 |
1 |
1 |
−1 |
−1 |
278 |
89.4 |
0.278 |
| 14 |
0 |
0 |
2 |
0 |
203 |
67.2 |
0.187 |
| 15 |
0 |
0 |
−2 |
0 |
264 |
83.1 |
0.262 |
| 16 |
−1 |
1 |
−1 |
−1 |
194 |
77.1 |
0.198 |
| 17 |
1 |
−1 |
1 |
1 |
229 |
87.2 |
0.276 |
| 18 |
1 |
−1 |
−1 |
1 |
261 |
94.7 |
0.332 |
| 19 |
−1 |
−1 |
−1 |
−1 |
238 |
82.6 |
0.256 |
| 20 |
0 |
0 |
0 |
0 |
204 |
74.5 |
0.221 |
| 21 |
0 |
0 |
0 |
0 |
197 |
77.5 |
0.227 |
| 22 |
0 |
2 |
0 |
0 |
171 |
69.7 |
0.181 |
| 23 |
−1 |
−1 |
1 |
1 |
183 |
62.5 |
0.196 |
| 24 |
0 |
0 |
0 |
0 |
203 |
73.4 |
0.228 |
| 25 |
−1 |
1 |
1 |
1 |
146 |
57.3 |
0.163 |
| 26 |
−1 |
1 |
1 |
−1 |
163 |
62.5 |
0.182 |
| 27 |
0 |
0 |
0 |
0 |
209 |
74.8 |
0.223 |
| 28 |
0 |
−2 |
0 |
0 |
238 |
81.7 |
0.282 |
| 29 |
1 |
−1 |
1 |
−1 |
273 |
92.7 |
0.323 |
| 30 |
1 |
1 |
1 |
−1 |
249 |
87.2 |
0.262 |
Higher r2 value and insignificant (p > 0.05) lack of fit showed good fit of regression model. PS, EE and PDI were obtained in the range of 146–324 nm, 57.3–95.7% and 0.163–0.394, respectively. Polymer content was found to be the dominating factor amongst all significant independent variables as evidenced by higher positive coefficient in regression analysis equations.
Eqn (4) explains the influence of different independent variables on PS of ALPNs which is depicted in Fig. 1A and B. Higher polymer content led to higher viscosity of organic phase which favored larger PS of ALPNs.38 TPGS concentration, volume of acetone and stirring speed possessed negative coefficient, indicating that an increment in these independent variables favor smaller PS. Increased TPGS concentration fortified boundary layer around particles, which limits the particles impingement and aggregation, resulting into mono dispersed nanoparticles with smaller PS.39 Increased volume of acetone lowered the viscosity of organic phase and favored smaller PS. Higher stirring speed favored smaller PS, as stirring provided shear force to break down the organic phase droplets.28
 |
| | Fig. 1 (A–F) Three dimensional response surface model showing the influence of independent variables X1, X2, X3 and X4 on particle size (A and B); entrapment efficiency (C and D) and polydispersity index (E and F). (X1: polymer weight, X2: TPGS concentration, X3: volume of acetone, X4: stirring speed, R1: hydrodynamic particle size, R2: entrapment efficiency and R3: polydispersity index.) | |
Eqn (5) as well as Fig. 1C and D demonstrate the effect of selected independent variables on EE of ALPNs. Raised polymer content favored enhancement of EE due to higher viscosity of organic phase, which have limited the drug diffusion from the organic phase subsequently.34,40 Negative coefficient of other variables indicated that an increase in these variables favor lowering of EE. An increment in TPGS concentration have increased the drug solubility in aqueous medium due to micelles formations and thus, reduced EE. Raised volume of acetone caused a reduction in organic phase viscosity, which led to lowering of EE. Higher stirring speed provided higher dispersing force, which led to facilitated diffusion of drug from organic phase to aqueous phase and resulted into lower EE.
Fig. 1E and F along with eqn (6) depict the influence of selected parameters on PDI of formulated ALPNs. Except polymer content, all other variables exhibited negative coefficient, which indicate an increase in these variables resulting into lessening of PDI. Zeta potential of ALPNs was found to be in the range of −18 to −29 mV. Negative potential was attributed to anionic nature of polymer and drug. Optimal values of independent formulation parameters were obtained as X1: 115 mg, X2: 0.8%, X3: 9 ml and X4: 1250 rpm with predicted response variables R1: 188 nm, R2: 78% and R3: 0.189 by setting the constraint minimum to PS and PDI as well as maximum to EE simultaneously in an optimization tool. Optimized batch of ALPNs was prepared by using the optimal formulation parameters and the observed response variables were 197 ± 5 nm PS, 75.6 ± 3.2% EE, 0.213 ± 0.012 PDI and −(24.4 ± 1.9) mV zeta potential, which were close to the predicted response variables. Reconstituted lyophilized sample of optimized ALPNs did not exhibit any significant change in PS, EE and PDI (Table 4). The optimized batch was subjected for further evaluation.
Table 4 Effect of lyophilisation on particle size, entrapment efficiency and PDI of ALPNsa
| ALPNs properties |
Before lyophilisation |
After lyophilisation with mannitol (2%) |
| Reconstituted lyophilized ALPNs in 1 ml of deionised water. (All results are shown as mean ± S.D., n = 3.) |
| Particle size (nm) |
197 ± 5 |
205 ± 6 |
| Entrapment efficiency (%) |
75.6 ± 3.2 |
75.2 ± 3.3 |
| PDI |
0.213 ± 0.012 |
0.221 ± 0.013 |
3.2 Solid state characterization
Fig. 2A illustrates FTIR spectra of ATR with characteristic bands at 3670 cm−1 (free O–H stretching of trihydrate), 3367 cm−1 (N–H stretching), 3222 cm−1 (asymmetric O–H stretching), 3055 cm−1 (symmetric O–H stretching), 1649 cm−1 (asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1578 cm−1 (symmetric CO stretching), 1550–1468 cm−1 (C–C ring stretching), 1317 cm−1 (CH3/CH2 deformation) and 1243 cm−1 (C–N stretching); which were retained in physical mixture as well as ALPNs.41 The carbonyl band of PCL was shifted from 1735 to 1737 cm−1 in placebo nanoparticles and ALPNs possibly due to overlapping of TPGS carbonyl band (1739 cm−1). The characteristics C–O–C stretching vibration of repeated –OCH2CH2 chain of TPGS from 1101 cm−1 to 1263 cm−1 were observed in ALPNs and placebo nanoparticles spectra, which suggest the presence of TPGS absorbed on the nanoparticles surface.42 Prominent peaks of placebo nanoparticles were also preserved in ALPNs. No any other peaks/bands were observed apart from ATR and placebo nanoparticles in ALPNs spectra which accorded good chemical compatibility of ATR in ALPNs.
O stretching), 1578 cm−1 (symmetric CO stretching), 1550–1468 cm−1 (C–C ring stretching), 1317 cm−1 (CH3/CH2 deformation) and 1243 cm−1 (C–N stretching); which were retained in physical mixture as well as ALPNs.41 The carbonyl band of PCL was shifted from 1735 to 1737 cm−1 in placebo nanoparticles and ALPNs possibly due to overlapping of TPGS carbonyl band (1739 cm−1). The characteristics C–O–C stretching vibration of repeated –OCH2CH2 chain of TPGS from 1101 cm−1 to 1263 cm−1 were observed in ALPNs and placebo nanoparticles spectra, which suggest the presence of TPGS absorbed on the nanoparticles surface.42 Prominent peaks of placebo nanoparticles were also preserved in ALPNs. No any other peaks/bands were observed apart from ATR and placebo nanoparticles in ALPNs spectra which accorded good chemical compatibility of ATR in ALPNs.
 |
| | Fig. 2 (A–F) Showing the (A) FTIR spectra, (B) DSC thermograms and (C) PXRD of atorvastatin, physical mixture, PCL placebo nanoparticles and ALPNs whereas, (D–F) showing atomic force microscopy, transmission electron microscopy and electron diffraction pattern of ALPNs, respectively. | |
Fig. 2B shows the DSC thermogram patterns of ATR, PM, PN and ALPNs. A broad endotherm ranging from 70–130 °C (could be attributed to loss of trihydrate) and a sharp endotherm at 165 °C (corresponded to melting point of ATR) was observed in ATR thermograph. These endotherms were also present in PM, which indicated good physical compatibility of drug with polymer. An endotherm in the region of 56–63 °C was noticed in each of PM, PNs and ALPNs which attributed to melting point of PCL. ATR loaded in ALPNs did not exhibit any endotherm at melting point conferred amorphous character of ATR distributed in ALPNs.
PXRD patterns of ATR, PM, PN and ALPNs are demonstrated in Fig. 2C. Many salient sharp diffraction peaks of ATR at 6.02, 9.06, 9.36, 10.16, 10.44, 11.76, 12.08, 16.92, 19.32, 21.50, 22.58, 23.18 and 23.56° of 2θ were observed and also remained in PM, which corroborated good physical compatibility of drug with polymer. No any prominent sharp peaks were noticed in PNs and ALPNs, which correspond to their amorphous character. ATR lost crystalline character in ALPNs probably due to smaller particle size and its dispersion into polymer matrix.
3.3 Morphology of ALPNs
Fig. 2D and E show AFM and TEM image of ALPNs, respectively. AFM image exhibited the discrete spherical shape with smooth surface of ALPNs and size is comparable to hydrodynamic PS. TEM micrograph also exhibited comparable PS of ALPNs to that of hydrodynamic PS. ED pattern of ALPNs, as shown in Fig. 2F, exhibited diffuse elastic scattered ring without any spotty diffraction patterns, confirming the amorphous character of ALPNs.43
3.4 In vitro release study
ATR release profile from the ALPNs illustrated initial burst release followed by subsequent sustained release (Fig. 3A). Fifty percent of drug was released in initial 2 h and completely released in 10 h from pure drug suspension. ALPNs released fifty percent of drug in 16 h while rest in 96 h. Release data of ALPNs were best fitted into KP model on the basis of higher correlation coefficient. The n value (<0.45) corresponded to Fickian diffusion controlled release mechanism.44 ALPNs exhibited 96 h sustained release profile of ATR with diffusion controlled mechanism.
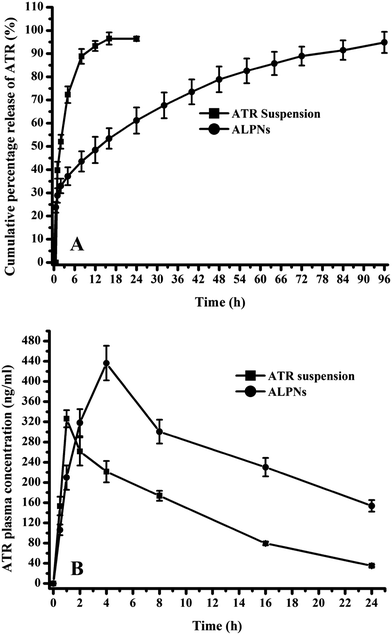 |
| | Fig. 3 Depicting the (A) in vitro atorvastatin release profile of ALPNs (B) pharmacokinetic profile of atorvastatin after single dose administration of drug suspension and ALPNs. (n = 6, dose: 20 mg kg−1 of atorvastatin and vertical bars represent the standard difference error.) | |
3.5 Stability study
3.5.1 Stability study in simulated biological fluid. The effect of different biological fluids on ALPNs properties (PS, EE and PDI) has been shown in Table 5. The change in PS and PDI value of ALPNs was observed to be insignificant (p > 0.05) in both simulated biological fluid (SGF and SIF) which indicated protection of ALPNs from aggregation and enzymatic degradation. Combined influence of zeta potential and stabilizer protect ALPNs from aggregation while polymer matrix and stabilizer provided protection of ALPNs from enzymatic degradation. Zeta potential, originated from electrostatic charge on ALPNs surface, repel each other and protect from aggregation. Stabilizer clung around ALPNs not only ensure stable dispersion but also provide physical barrier against enzymatic degradation of ALPNs and drug. EE was found to be slightly changed after incubation in SIF of pH 6.8 whereas; EE was remained same in SGF of pH 1.2 which were attributed to the pH dependent solubility of ATR. ATR solubility increases with the increase of pH of media. Study exhibited that the ALPNs possessed good physical stability with slight change in EE against gastro-intestinal milieu.
Table 5 Stability of ALPNs in different biological fluida
| ALPNs property |
Before incubation |
After incubation in SGF pH 1.2 |
After incubation in SIF pH 6.8 |
| Time (2 h) |
Time (6 h) |
| PS: particle size, EE: entrapment efficiency, PDI: polydispersity index. (All results are shown as mean ± S.D., n = 3.) |
| PS (nm) |
197 ± 5 |
203 ± 6 |
201 ± 4 |
| EE (%) |
75.6 ± 3.2 |
75.2 ± 3.7 |
69.2 ± 4.3 |
| PDI |
0.213 ± 0.012 |
0.227 ± 0.015 |
0.217 ± 0.013 |
3.5.2 Storage stability study. Storage stability study was incorporated to assess the potential of ALPNs to withstand the environmental changes. Physical appearance of ALPNs did not change in different environmental conditions. The properties of ALPNs stored under different environmental conditions have been depicted in Fig. 4. A significant change (p < 0.05) in ALPNs properties (PS, EE and PDI) was observed after storage under accelerated condition which was attributed to instability of ALPNs due to agglomeration of nanoparticles and degradation of polymer along with drug. However, insignificant change (p > 0.05) in ALPNs properties was observed after storage under normal condition (25 ± 2 °C/60 ± 5% RH) and refrigerated condition (4 ± 1 °C). Thus, ALPNS should be stored below room temperature (<25 °C), preferably at refrigerated condition for long term use.
 |
| | Fig. 4 The particle size, entrapment efficiency and polydispersity index (PDI) of optimized ALPNs during stability study of 6 months under different environmental conditions [* represents a significant change (p < 0.05) compared to properties of ALPNs on 0 days]. | |
3.6 Pharmacokinetic study
Rat plasma ATR concentration time profiles after single oral dose administration of DS and ALPNs are shown in Fig. 3B. Different pharmacokinetic parameters of DS and ALPNs are summarized in Table 6. Two times enhancement in AUC0–24 h and 2.8 times in AUC0–∞ were noticed in ALPNs group in comparison to DS group. The significant enhancement in bioavailability attributed to enhanced absorption of ATR incorporated in ALPNs, which might be due to smaller particle size, higher uptake of nanoparticles via Payer's patches and TPGS′ bioavailability enhancing properties.45,46 A significant enhancement (1.34 times) in maximum plasma concentration (Cmax) was observed in ALPNs due to higher absorption of ALPNs from GIT. Tmax was observed to be higher in ALPNs (4 h) than the DS (1 h), which might be attributed to sustained drug release features of ALPNs. Mean residence time (MRT) of ATR was detected significantly higher in ALPNs, probably due to sustained drug release behaviour and surface absorbed TPGS of ALPNs, which might have improved the residence period, possibly by providing hydrophilic surface (PEG of TPGS) to nanoparticles.30,46 Hence, ALPNs provided a superior nanoparticles platform in order to enhance the oral bioavailability.
Table 6 Pharmacokinetic parameters of atorvastatin calcium after single dose oral administration of ALPNs and atorvastatin suspension (n = 6 and dose: 20 m kg−1 of atorvastatin)a
| Pharmacokinetic parameter |
ALPNs |
Atorvastatin calcium suspension |
| Data are represented as mean ± SEM. Statistical significance compared with atorvastatin suspension. *p < 0.05, **p < 0.01 and ***p < 0.001. ALPNs: atorvastatin loaded PCL nanoparticles. |
| Cmax (ng ml−1) |
436.2 ± 15.3* |
326.2 ± 6.9 |
| Tmax (h) |
4.0 |
1.0 |
| AUC0–24 h (ng h ml−1) |
6204.0 ± 205.2** |
3113.0 ± 55.8 |
| AUC0–∞ (ng h ml−1) |
9754.7 ± 309.2** |
3470.9 ± 58.7 |
| AUMCtotal (ng h2 ml−1) |
222467 ± 7112*** |
38368 ± 619 |
| MRT (h) |
23.88 ± 0.85*** |
11.06 ± 0.15 |
3.7 Efficacy and safety study
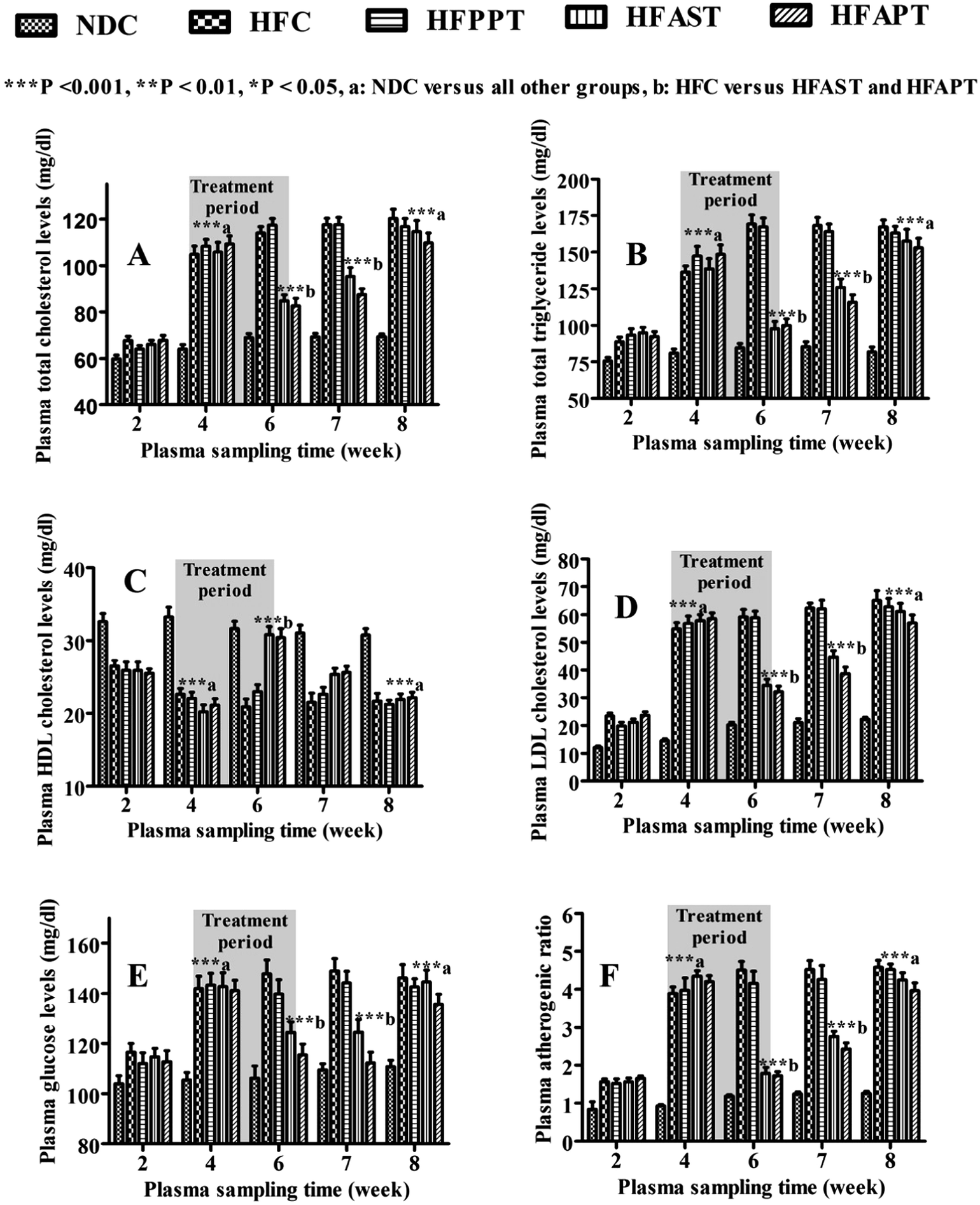
A significant elevation in plasma total cholesterol (75–81%), triglyceride (75–84%), low density lipoprotein-cholesterol (200–300%), glucose level (32–39%) and atherogenic index (318–367%) while a significant reduction in high density lipoprotein-cholesterol (27–33%) were noticed in high fat-diet rat groups than the NDC group, at the end of 4th week (hyperlipidemia induction period) (Fig. 5). Different groups were treated with different regimen as mentioned in Table 2 during treatment period (4th–6th week). Dose size of ALPNs to HFAPT group was chosen half of equivalent weight of pure ATR administered to HFAST group on the basis of pharmacokinetic output. The shaded region in Fig. 5 indicates the treatment period during fourth to sixth week. At the end of sixth week, a significant reduction in elevated level of PTC, PTG, LDL-C, VLDL-C and glucose were noticed in HFAST and HFAPT groups (Fig. 5 and Table 7). Plasma HDL-C level was found to be significantly improved in ATR and ALPNs treated groups at the end of sixth week. The plasma AI was found to be significantly decreased in HFAPT and HFAST group than HFC and HFPPT groups. The pattern of elevation or reduction in plasma lipid profile was approximately similar in HFAPT and HFAST group during treatment period. During 6th–7th week, the plasma lipid elevation rates were observed to be slightly slower in HFAPT group than the HFAST group, which might be attributed to sustained release of drug from ALPNs. No significant difference was observed between plasma lipid and glucose profile of HFC and HFPPT group, concluding insignificant effect of placebo nanoparticles itself on efficacy profile. At the end of 8th week, all the high fat diet groups exhibited similar plasma lipid and glucose profile, indicating drug washed out completely during 6th to 8th week. Efficacy study exhibited similar profile during treatment (4th–6th week) and drug wash out (6th–8th week) period in both the HFAST and HFAPT group, which showed dosage regimen of ALPNs containing 1.5 mg of ATR was equally efficacious to that of ATR suspension containing 3 mg of ATR. The prominent outcome of this study was that ALPNs at half dose of ATR were equally effective to that of ATR suspension.
 |
| | Fig. 5 (A–F) Showing the plasma (A) total cholesterol, (B) triglyceride, (C) HDL cholesterol, (D) LDL cholesterol, (E) glucose and (F) atherogenic ratio level time profile of different groups. (Error bars showing S.E.M. and Bonferroni post test two way ANOVA statistics applied.) NDC: normal diet control, HFC: high fat diet control, HFPPT: high fat-diet PCL-placebo-nanoparticles treated, HFAST: high fat-diet atorvastatin suspension treated, HFAPT: high fat-diet ALPNs treated, HDL: high density lipoprotein and LDL: low density lipoprotein ***p < 0.001, (a) statistically significant versus NDC, (b) statistically significant versus HFC and HFPPT within same week. | |
Table 7 Biochemical efficacy and safety parameters of plasma samples collected from different groups at the end of 6th weeka
| S. No. |
Biochemical parameters |
NDC |
HFD |
HFPPT |
HFAST |
HFAPT |
| PTC: plasma total cholesterol; PTG: plasma triglyceride; VLDL-C: very low density lipoprotein cholesterol; HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; PG: plasma glucose; AI: plasma atherogenic index; BUN blood urea nitrogen; CK: creatinine kinase; LDH: lactate dehydrogenase; AST: aspartate transaminase; NDC: normal diet controlled; HFC: high fat-diet controlled; HFPPT: high fat-diet placebo-PCL-nanoparticles treated; HFAST: high fat-diet atorvastatin calcium suspension treated; HFAPT, high fat-diet ALPNs treated. All values are expressed in mean ± SEM (n = 6) ***p < 0.001, **p < 0.01 and *p < 0.05. aStatistically significant versus NDC. bStatistically significant versus HFC. cStatistically significant versus NDC, HFC and HFPET. dStatistically significant versus HFAST. |
| 1 |
PTC (mg dl−1) |
68.9 ± 1.7 |
113.9 ± 2.7***a |
117.3 ± 2.9***a |
84.9 ± 2.5***b |
82.6 ± 3.2***b |
| 2 |
PTG (mg dl−1) |
84.6 ± 3.0 |
169.3 ± 6.2***a |
167.2 ± 6.1***a |
97.7 ± 5.0***b |
99.9 ± 4.6***b |
| 3 |
VLDL-C (mg dl−1) |
16.9 ± 0.6 |
33.9 ± 1.2***a |
33.7 ± 1.2***a |
19.4 ± 1.0***b |
20.0 ± 0.9***b |
| 4 |
HDL-C (mg dl−1) |
31.7 ± 1.0 |
20.9 ± 1.0***a |
23.0 ± 0.91***a |
30.8 ± 1.1***b |
30.4 ± 1.2***b |
| 5 |
LDL-C (mg dl−1) |
20.3 ± 0.9 |
59.2 ± 2.7***a |
58.8 ± 2.4***a |
34.6 ± 2.2***b |
32.2 ± 2.0***b |
| 6 |
PG (mg dl−1) |
106.2 ± 4.9 |
147.8 ± 5.5***a |
139.7 ± 5.7***a |
124.4 ± 4.2**b |
115.5 ± 4.3***b |
| 7 |
AI |
1.18 ± 0.04 |
4.50 ± 0.23***a |
4.16 ± 0.32***a |
1.78 ± 0.17 |
1.73 ± 0.1 |
| 8 |
Plasma creatinine level (mg dl−1) |
1.25 ± 0.06 |
1.57 ± 0.05 |
1.44 ± 0.06 |
3.67 ± 0.26***c |
1.82 ± 0.08***d |
| 9 |
BUN (mg dl−1) |
13.9 ± 0.6 |
14.1 ± 0.6 |
14.6 ± 0.7 |
30.6 ± 2.3***c |
16.2 ± 0.74***d |
| 10 |
CK (U l−1) |
117.3 ± 7.3 |
112.7 ± 11.7 |
139.7 ± 9.7 |
128.5 ± 8.4 |
131.5 ± 10.6 |
| 11 |
LDH (U l−1) |
244.8 ± 28.1 |
246.7 ± 26.3 |
235.8 ± 16.1 |
451.2 ± 41.5***c |
270.7 ± 19.0***d |
| 12 |
AST (U l−1) |
42.3 ± 2.0 |
45.1 ± 2.3 |
50.7 ± 2.6 |
66.8 ± 3***c |
48.2 ± 2.0***d |
Long term therapy of escalated dose of statins may cause severe skeleton muscle toxicity like rhabdomyolysis.47 Rhabdomyolysis can be diagnosed by examining plasma CK profile and creatinine level. Plasma CK level greater than ten times of upper limit of normal CK level with elevated creatinine level can clearly indicate the pathogenesis of rhabdomyolysis.48 Elevated BUN, LDH and AST level apparently show manifestation of membrane damage.49 Safety study of ATR and ALPNs were assessed by determining the plasma biochemical parameters (creatinine, BUN, CK, LDH and AST levels) of collected plasma samples at 4th, 6th and 8th week. Muscle toxicity can be detected and diagnosed by assessing above mentioned parameters. Plasma CK data of all the animal groups were found within normal range. Plasma creatinine, BUN, LDH and AST levels of HFAST group were diagnosed significantly higher than other groups at the end of 6th week, which clearly indicated mild muscle toxicity and absence of rhabdomyolysis (Table 7). These four biochemical parameters of HFAPT group were observed to be similar or slightly elevated than the NDC, HFC and HFPPT; which concluded that ALPNs elicited negligible muscle toxicity (Fig. 6 and Table 7). ALPNs treated group exhibited a significant improvement in safety parameters than the pure ATR treated group. The reduced toxicity of ALPNs might be attributed to reduced dose size of ATR, sustained drug delivery action, protection of drug from degradation in GIT milieu, constrained drug in vascular system and limited exposure of drug to muscular system. Histology of liver tissue of different groups exhibited normal rubicund cells which inferred no toxic effect of ATR and ALPNs on liver (Fig. 7).
 |
| | Fig. 6 Showing plasma (A) creatinine levels (B) blood urea nitrogen (BUN) levels (C) creatinine kinase (CK) levels, (D) plasma lactate dehydrogenase (LDH) levels and (E) aspartate amino transferase (AST) levels versus time profile in different groups. (Error bars represent S.E.M. and Bonferroni post test two way ANOVA statistics has been applied.) NDC: normal diet control, HFC: high fat diet control, HFPPT: high fat-diet PCL-placebo-nanoparticles treated, HFAST: high fat-diet atorvastatin suspension treated, HFAPT: high fat-diet ALPNs treated. | |
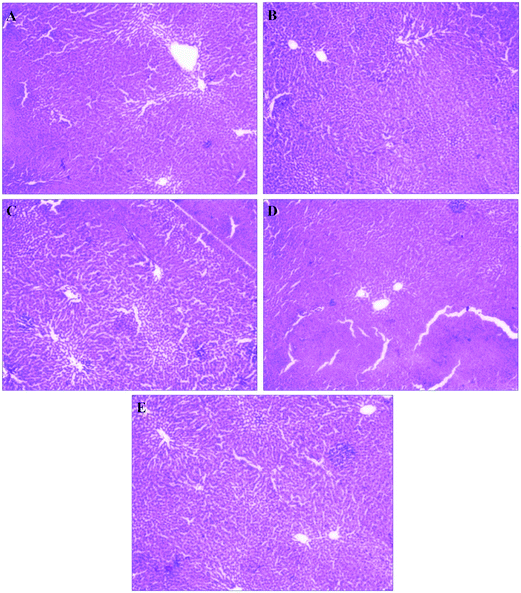 |
| | Fig. 7 Histomicrograph of stained liver tissue of (A) normal diet control, (B) high fat-diet control (C) placebo nanoparticle treated, (D) ATR suspension treated and (E) ALPNs treated groups. | |
4. Conclusion
In this study, atorvastatin calcium loaded poly(ε-caprolactone) nanoparticles (ALPNs) were successfully prepared, optimized and evaluated for its physicochemical, morphological properties, in vitro release, stability, in vivo pharmacokinetic, efficacy and safety profiles. Physicochemical and morphological assessment exhibited interaction free stable homogeneously distributed spherical nanoparticles. The outcome of pharmacokinetic and biochemical parameters assessment showed the ALPNs were more effective and safer than pure ATR suspension for the treatment of atherosclerosis as well as hyperlipidemia in experimental rats. The study corroborated with the proposed hypothesis of ALPNs as a promising novel drug delivery system for sustained release with enhanced bioavailability, efficacy and safety profile of ATR.
Declaration of conflicts of interest
Authors declare no conflicts of interest.
Acknowledgements
First author is thankful to ministry of human resource development, New Delhi, for financial assistance as teaching assistantship for carrying out this research work. All authors would like to acknowledge respective Heads of Physics and Chemistry department, Banaras Hindu University, Varanasi, for providing facility of DSC, PXRD, TEM and AFM respectively.
References
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS PubMed.
- B. Daglar, E. Ozgur and M. E. Corman, et al., RSC Adv., 2014, 4, 48639–48659 RSC.
- R. Juneja and I. Roy, RSC Adv., 2014, 4, 44472–44479 RSC.
- Y. B. Patil, U. S. Toti and A. Khdair, et al., Biomaterials, 2009, 30, 859–866 CrossRef CAS PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef PubMed.
- A. Kowalczuk, R. Trzcinska and B. Trzebicka, et al., Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS.
- N. K. Swarnakar, A. K. Jain and R. P. Singh, et al., Biomaterials, 2011, 32, 6860–6874 CrossRef CAS PubMed.
- C. K. Glass and J. L. Witztum, Cell, 2001, 104, 503–516 CrossRef CAS PubMed.
- C. B. Xu, E. Stenman and L. Edvinsson, Biochem. Pharmacol., 2002, 64, 497–505 CrossRef CAS PubMed.
- M. Labiós, M. Martínez and F. Gabriel, et al., Thromb. Res., 2005, 115, 263–270 CrossRef PubMed.
- F. Violi, R. Carnevale, D. Pastori and P. Pignatelli, Trends Cardiovasc. Med., 2014, 24, 142–148 CrossRef CAS PubMed.
- D. C. Souza-Costa, V. C. Sandrim and L. F. Lopes, et al., Atherosclerosis, 2007, 193, 438–444 CrossRef CAS PubMed.
- P. Xu, H. Yu and Z. Zhang, et al., Biomaterials, 2014, 35, 7574–7587 CrossRef CAS PubMed.
- H. Lennernas, Clin. Pharmacokinet., 2003, 42, 1141–1160 CrossRef PubMed.
- M. Takano, R. Yumoto and T. Murakami, Pharmacol. Ther., 2006, 109, 137–161 CrossRef CAS PubMed.
- C. N. Magee, S. A. Medani and S. F. Leavey, et al., Am. J. Kidney Dis., 2010, 56, 11–15 CrossRef PubMed.
- S. Khan, S. Baboota, J. Ali, R. S. Narang and J. K. Narang, Drug Dev. Ind. Pharm., 2015, 1–12, DOI:10.3109/03639045.2015.1040414.
- K. A. Ali, B. Mukherjee and A. K. Bandyopadhyay, Drug Dev. Ind. Pharm., 2013, 39, 1742–1749 CrossRef CAS PubMed.
- A. G. Agrawal, A. Kumar and P. S. Gide, Drug Dev. Ind. Pharm., 2015, 41, 594–604 CrossRef CAS PubMed.
- R. N. Kishore, P. R. Yalavarthi, H. C. Vadlamudi, K. R. Vandana, A. Rasheed and M. Sushma, Drug Dev. Ind. Pharm., 2015, 41, 1213–1222 CrossRef PubMed.
- M. Bandivadekar, S. Pancholi, R. Kaul-Ghanekar, A. Choudhari and S. Koppikar, Drug Dev. Ind. Pharm., 2013, 39, 696–703 CrossRef CAS PubMed.
- H. Shen and M. Zhong, J. Pharm. Pharmacol., 2006, 58, 1183–1191 CrossRef CAS PubMed.
- M. S. Kim, J. S. Kim and W. Cho, et al., Int. J. Biol. Macromol., 2013, 59, 138–142 CrossRef CAS PubMed.
- J. S. Kim, M. S. Kim and H. J. Park, et al., Int. J. Pharm., 2008, 359, 211–219 CrossRef CAS PubMed.
- M. S. Kim, S. J. Jin and J. S. Kim, et al., Eur. J. Pharm. Biopharm., 2008, 69, 454–465 CrossRef CAS PubMed.
- M. Anwar, M. H. Warsi and N. Mallick, et al., Eur. J. Pharm. Sci., 2011, 44, 241–249 CrossRef CAS PubMed.
- N. Kumar, S. Chaurasia and R. R. Patel, et al., Adv. Sci. Lett., 2014, 20, 984–993 CrossRef.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- P. R. Vuddanda, V. M. Rajamanickam, M. Yaspal and S. Singh, BioMed Res. Int., 2014, 951942, DOI:10.1155/2014/951942.
- P. Chaubey, R. R. Patel and B. Mishra, Expert Opin. Drug Delivery, 2014, 11, 1163–1181 CrossRef CAS PubMed.
- R. Chawla, S. Jaiswal and B. Mishra, Expert Opin. Drug Delivery, 2014, 11, 31–43 CrossRef CAS PubMed.
- J. Hao, F. Wang and X. Wang, et al., Eur. J. Pharm. Sci., 2012, 47, 497–505 CrossRef CAS PubMed.
- R. R. Patel, G. Khan, S. Chaurasia, N. Kumar and B. Mishra, RSC Adv., 2015, 5, 76491–76506 RSC.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- A. K. Jain, N. K. Swarnakar, C. Godugu, R. P. Singh and S. Jain, Biomaterials, 2011, 32, 503–515 CrossRef CAS PubMed.
- Y. Hou, W. Shao and R. Xiao, et al., Exp. Gerontol., 2009, 44, 434–439 CrossRef PubMed.
- S. Galindo-Rodriguez, E. Allémann, H. Fessi and E. Doelker, Pharm. Res., 2004, 21, 1428–1439 CrossRef CAS.
- Y. Wang, Y. Zheng and L. Zhang, et al., J. Controlled Release, 2013, 172, 1126–1141 CrossRef CAS PubMed.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83, 389–400 CrossRef CAS PubMed.
- H. X. Zhang, J. X. Wang and Z. B. Zhang, et al., Int. J. Pharm., 2009, 374, 106–113 CrossRef CAS PubMed.
- Y. Zheng, H. Chen and X. Zeng, et al., Nanoscale Res. Lett., 2013, 161, 1–12, DOI:10.1186/1556-276X-8-161.
- H. Stormer, H. J. Kleebe and G. Ziegler, J. Non-Cryst. Solids, 2007, 353, 2867–2877 CrossRef.
- Z. Fan, J. Wu, X. Fang and X. Sha, Int. J. Pharm., 2013, 445, 141–147 CrossRef CAS PubMed.
- Z. Zhang, S. Tan and S. S. Feng, Biomaterials, 2012, 33, 4889–4906 CrossRef CAS PubMed.
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed.
- M. J. Cziraky, V. J. Willey and J. M. McKenney, et al., J. Clin. Lipidol., 2013, 71, 102–108 CrossRef PubMed.
- A. Holbrook, M. Wright and M. Sung, et al., Can. J. Cardiol., 2011, 27, 146–151 CrossRef CAS PubMed.
- G. D. Giannoglou, Y. S. Chatzizisis and G. Misirli, Eur. J. Intern. Med., 2007, 18, 90–100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 shows the release kinetic rate constant, correlation coefficient and other parameters of various drug release kinetic models. Fig. S1 depicts gas chromatograms of ALPNs aqueous sample and standard acetone sample. Fig. S2 exhibits the drug release profile of ALPNs in simulated biological fluids (SGF and SIF). See DOI: 10.1039/c5ra26674b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.